




Sellar masses are a heterogeneous group of lesions, both in nature and management. Not all of them require surgery.
ObjectivesTo describe the presenting symptoms of sellar masses and endocrine abnormalities occurring during follow-up. To emphasize the significance of endocrine assessment, and to identify lesions amenable to hormonal treatment.
Patients and methodsA retrospective review of the records of children under 14 years of age referred to our center for sellar lesions during a period of 12 years. Data collected included sex, age, nature of lesion, clinical presentation, size, treatment, and endocrine abnormalities.
ResultsForty-five patients (25 females) aged 7.2±4.1 years (range 0.25–13.5) were enrolled. Follow-up time was 6.2±3.7 years. Lesion nature was known in 39 cases, 4 of which were successfully treated at the Endocrinology Department: 3 prolactinomas (with dopamine agonist) and one thyrotroph cell hyperplasia (with levothyroxine). The most common presenting symptoms were neurological and/or visual (25/45), followed by endocrine conditions (13/45). Duration of endocrine and neuro-ophthalmic symptoms was 12.6±18.2 months and 2.6±4.9 months (p=0.012), respectively. Some endocrine conditions were found in 24/45 patients at the initial evaluation and in 37/45 patients at the end of follow-up.
ConclusionsManagement of sellar lesions requires a multidisciplinary effort. Endocrine tests are indispensable to identify lesions amenable to hormonal treatment. Endocrine disorders usually occurred before neurological and ophthalmological symptoms, and their identification may therefore allow for earlier diagnosis. Hormone assessment should be regularly performed during follow-up.
Las lesiones selares son heterogéneas en su naturaleza y abordaje. No todas requieren cirugía.
ObjetivosDescribir las formas de presentación de las lesiones selares y la presencia de endocrinopatías a lo largo de la evolución, así como resaltar la importancia de la valoración endocrinológica e identificar aquellas lesiones subsidiarias de tratamiento hormonal.
Pacientes y métodos: Estudio retrospectivo de los niños menores de 14 años remitidos a nuestro centro por lesiones selares durante 12 años. Se recogieron las siguientes variables: sexo, edad, naturaleza de la lesión, presentación clínica, tamaño, tratamiento primario y presencia de endocrinopatías.
ResultadosSe incluyen 45 pacientes (25 mujeres) con edades comprendidas desde los 3 meses hasta 13,5 años (media 7,2±4,1) y un tiempo de seguimiento de 6,2±3,7 años. Se conoce la naturaleza de la lesión en 39 casos, de los cuales 4 se han tratado eficazmente por Endocrinología: 3 prolactinomas con cabergolina, y una hiperplasia hipofisaria con levotiroxina. El motivo de consulta fueron síntomas de neuropatía y oftalmopatía en 25/45 casos, y de endocrinopatía en 13/45. El periodo de síntomas endocrinológicos previos fue de 12,6±18,2 meses, frente a 2,6±4,9 meses de los neurooftalmológicos (p=0,012). En el momento del diagnóstico, 24/45 pacientes presentaban alguna endocrinopatía, ascendiendo a 37/45 al final del seguimiento.
ConclusionesLas lesiones del área selar requieren un abordaje multidisciplinario. El estudio endocrinológico es imprescindible para identificar las que son subsidiarias de tratamiento hormonal. Los síntomas o signos de endocrinopatía pueden aparecer antes que los de neuropatía u oftalmopatía, por lo que es fundamental reconocerlos para adelantar el diagnóstico. La evaluación hormonal debe repetirse periódicamente a lo largo de la evolución.
Lesions in the sellar area (hypothalamic-pituitary-chiasmatic) are uncommon in children and adolescents (only 6% of them occur in these age groups) and highly heterogeneous in nature and presentation.1,2 Cases are different as compared to adults,3 and adequate differential diagnosis is required, because therapeutic management is very different depending on etiology.
Craniopharyngioma is the most common lesion in the first two decades of life, and should be treated with surgery. Prolactinoma is rare at these ages, but this does not mean that it should not be considered. Both prolactinoma and pituitary hyperplasia require medical treatment. Surgery is also not required for other lesions in this area, such as germinal tumors, for which chemotherapy and radiation therapy are the treatment of choice, and optic tract gliomas, which often require no treatment at all.4–6
Although most lesions are histologically benign, they are associated to high morbidity rates because of their proximity to vital structures. They are usually diagnosed based on visual or neurological changes caused by a mass effect. In addition, they are associated to endocrine conditions including hypothalamic and pituitary disease due to either hyposecretion or hypersecretion of some hormone. These are very often the first manifestations of the lesion.2,7
Our study objectives are to describe the presentation forms of sellar area lesions in children and adolescents, to analyze the presence of endocrine diseases at presentation and during their course, and to emphasize the importance of endocrine assessment, identifying lesions amenable to hormone therapy.
Patients and methodsA retrospective study was conducted of patients under 14 years of age referred to the pediatric endocrinology unit of a tertiary hospital for a sellar mass during a 12-year period (2000–2011). Lesions had been diagnosed by magnetic resonance imaging (MRI).
The following variables were collected from the clinical history of each patient: sex, age at diagnosis, nature of the lesion, clinical presentation symptoms and their onset time, diameter of the longest axis of the mass at diagnosis and in MRI, primary treatment, and presence of endocrine diseases at diagnosis and during the follow-up period.
Clinically presented symptoms and signs were defined as those that were first related to the lesion leading to seek medical help, but all signs and symptoms reported or seen before diagnosis were also collected. Presentation forms were categorized depending on whether signs and symptoms were due to neuropathy (headache, signs of cranial hypertension, seizures, focal signs), ophthalmopathy (decreased acuity or visual field, strabism, nystagmus), or endocrine diseases. The prior period with clinical signs and symptoms was defined as time from the start of symptoms attributable to the lesion (even if they did not lead to seek medical help) to lesion diagnosis.
Endocrine diseases included those of hypothalamic origin (obesity and eating, thirst, sleep or temperature disorders) and pituitary conditions due to hyposecretion of any hormone from the anterior pituitary gland (growth hormone [GH] deficiency, hypocortisolism, central hypothyroidism and hypogonadism) or the posterior pituitary gland (central diabetes insipidus), or due to hormone hypersecretion (hyperprolactinemia, early puberty, and gigantism). To diagnose obesity, age- and sex-specific thresholds of body mass index were used. Standard of care clinical and laboratory assessments were performed in the patients at lesion diagnosis (except in three patients with craniopharyngioma, prolactinoma, and dermoid cyst respectively, in whom they were performed after surgical resection) and every six months thereafter.
Quantitative variables (age and time periods) were given as mean±standard deviation. Prior periods with neuro-ophthalmological and endocrine clinical signs were compared using a Wilcoxon test, and the proportions of patients with endocrine disease in each group by type of lesion were compared using a Chi-square test.
ResultsForty-five patients (25 females) with age at diagnosis ranging from 3 months to 13.5 years (mean 7.2±4.1 years) and a follow-up time of 6.2±3.7 years were enrolled in the study. The greater lesion diameter was longer than 20mm in 29 patients. Lesion nature was known in 39 patients: 13 optic tract gliomas (5 in patients previously diagnosed with neurofibromatosis type 1), 10 germinal tumors, 6 craniopharyngiomas, 4 prolactinomas, 4 histiocytic granulomas, one dermoid cyst, and a case of pituitary hyperplasia secondary to congenital primary hyperparathyroidism diagnosed very late, at 9 years of age. Table 1 shows the variables at diagnosis in the total cohort and by type of lesion.
Variables of sellar lesions at diagnosis in the complete cohort and by type.
| All | Gliomas | Germinal tumors | Craniopharyngiomas | Prolactinomas | Histiocytomas | Undiagnosed | |
| No. | 45 | 13 | 10 | 6 | 4 | 4 | 6 |
| Females | 25 | 6 | 10 | 1 | 2 | 1 | 3 |
| Age at diagnosis (years) | 7.2±4.1 | 2.8±2.3 | 9.4±1.7 | 7.2±3.3 | 13.2±0.3 | 7.0±4.1 | |
| Prior months with endocrine symptoms | 12.6±18.2 | 7.0±4.6 | 25.0±26.2 | 9.6±10.0 | 0.7±0.3 | 4.2±2.1 | |
| Prior months with symptoms of neuropathy or ophthalmopathy | 2.7±4.9 | 1.1±1.3 | 2.0±2.1 | 1.5±1.0 | 15.0±12.7 | ||
| Years of follow-up | 6.2±3.7 | 7.9±3.3 | 5.5±3.0 | 7.1±5.1 | 3.2±2.0 | 5.5±5.9 | 6.0±2.4 |
| Form of presentation | |||||||
| Clinical signs of neuro-ophthalmopathy | 25 | 9 | 7 | 6 | 2 | 0 | 0 |
| Subclinical (neuroimaging) | 7 | 2 | 0 | 0 | 0 | 0 | 5 |
| Clinical signs of endocrine disease | 24 | 3 | 10 | 3 | 2 | 4 | 1 |
| As reason for consultation | 13 | 2 | 3 | 0 | 2 | 4 | 1 |
| At directed history | 11 | 1 | 7 | 3 | 0 | 0 | 0 |
| Early puberty | 4 | 3 | 0 | 0 | 0 | 0 | 1 |
| Diabetes insipidus | 14 | 0 | 9 | 2 | 0 | 3 | 0 |
| Galactorrhea | 2 | 0 | 0 | 0 | 2 | 0 | 0 |
| Obesity | 2 | 0 | 0 | 1 | 0 | 1 | 0 |
| Low height | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Anorexia | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Diameter>20mm by neuroimaging | 1 | 0 | 7 | 6 | 2 | 0 | 0 |
| Treatment for sellar lesion | |||||||
| Surgery | 9 | 2 | 0 | 5 | 1 | 0 | 0 |
| Chemotherapy | 6 | 3 | 0 | 0 | 0 | 3 | 0 |
| Chemotherapy and radiotherapy | 1 | 1 | 10 | 0 | 0 | 0 | 0 |
| Surgery and chemotherapy | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Surgery and radiotherapy | 1 | 0 | 0 | 1 | 0 | 0 | 0 |
| Cabergoline | 3 | 0 | 0 | 0 | 3 | 0 | 0 |
| Levothyroxine | 1 | ||||||
| None | 13 | 6 | 0 | 0 | 0 | 1 | 6 |
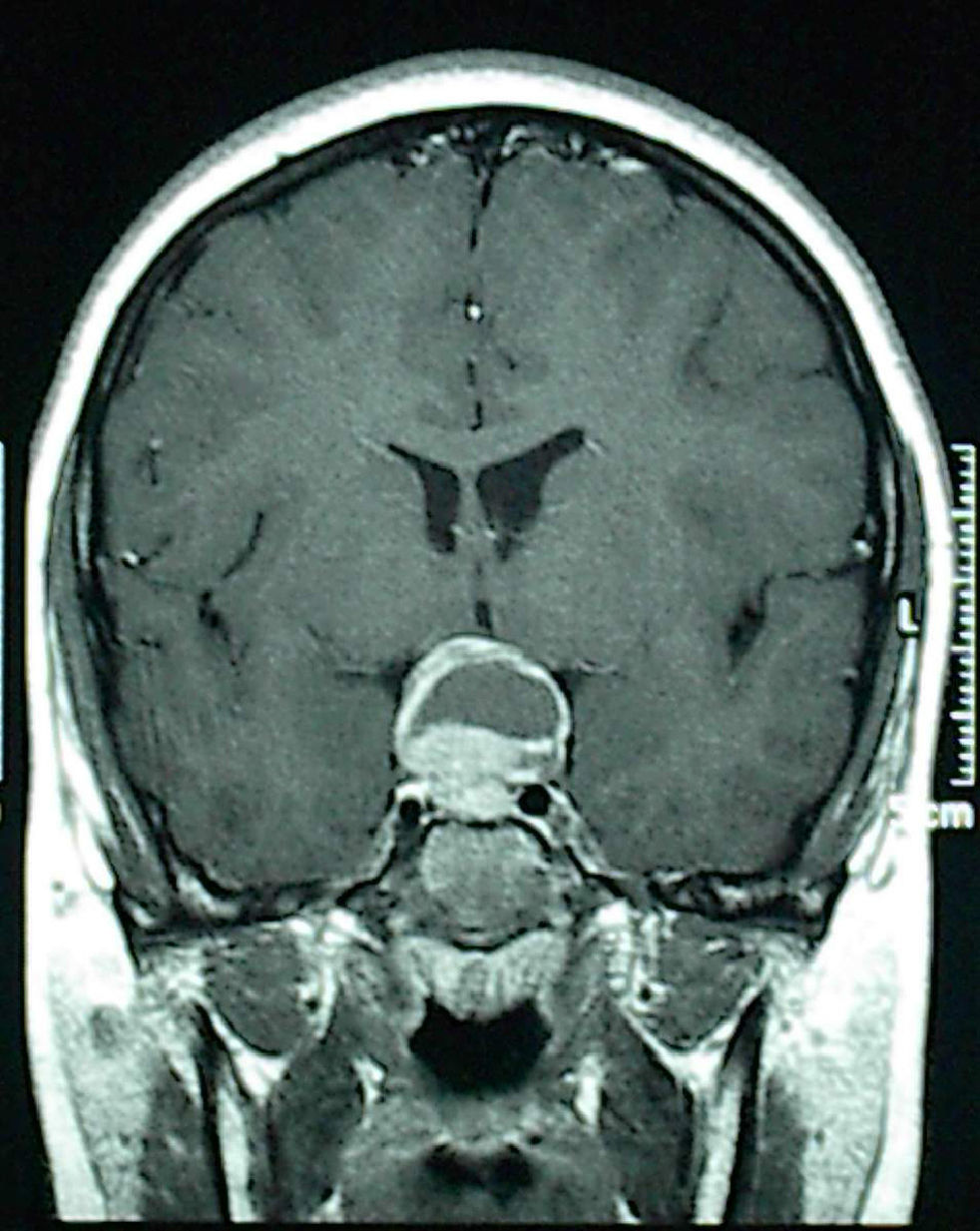
Four patients were successfully treated at the endocrinology department: 3 prolactinomas with the dopamine agonist cabergoline, while the patient with pituitary hyperplasia received levothyroxine. A 13-year-old boy complaining of headache underwent surgery with no prior hormone testing because neuroimaging revealed a lesion suspected to be craniopharyngioma because of its cystic appearance and large suprasellar extension (Fig. 1). The pathological laboratory diagnosed prolactinoma, and the cystic area was due to tumor apoplexy. The three patients with medically treated prolactinomas, all of them aged 13 years, were two pubertal girls complaining of isolated galactorrhea and with adenomas less than 10mm in size, and a boy reporting headache for the past few months and who had another cystic macroadenoma. Medical history of males included no evidence of endocrine disease that could have allowed for earlier diagnosis; they had not started puberty, and one of them showed mild bilateral gynecomastia 15mm in diameter, which could be considered physiological at this age.

In addition to the four patients with hormone treatment alone, oncological management (chemotherapy and/or radiotherapy) was decided in 17 children, and 13 children required no treatment for the lesion (they had no clinical signs and remained stable, as shown by regular neuroimaging tests). Only 11 patients required surgery for the mass, and two of these subsequently needed chemotherapy and radiotherapy. Hormone testing also revealed four tumors secreting chorionic gonadotropin. Diagnosis of germinoma was therefore established without the need for biopsy, and these patients were directly referred to the oncology department with no prior surgery.
Reasons for consultation were that the symptoms related to neuropathy and ophthalmopathy in 25 patients, and symptoms were related to endocrine disease in 13 patients: diabetes insipidus in 6, early puberty in 3, galactorrhea in 2, low growth in one, and weight gain in one. Seven lesions were subclinically diagnosed by neuroimaging, including 2 in workup study for neurofibromatosis type 1 and 5 that may be considered as incidentalomas, because problems not related to sellar disease (epilepsy, psychomotor retardation, syncope) were being investigated. It should be noted that 11 patients complaining of neurological or visual clinical signs and symptoms reported in the directed history long-standing endocrine clinical signs not previously assessed (8 of them severe polydipsia/polyuria for 1–5 years). Mean duration of prior endocrine symptoms was 12.6±18.2 months, longer than duration of neuro-ophthalmological symptoms, which was 2.6±4.9 months (p=0.012).
Half of the 24 patients with endocrine disorders at diagnosis (13 patients in whom they led to consultation and 11 patients in whom they were discovered by directed history) had more than one endocrine disorder. The proportion of patients with some hormone change was 30/45 at one year and 37/45 at the end of the follow-up period (Table 2). Presence of endocrine disease at diagnosis and in the first year of its course was related to nature of the lesion, but not to any other variable (age, sex, tumor size); optic tract gliomas had less endocrine changes at diagnosis (p=0.004) and at one year (p<0.001), 3/13 in both evaluations, consisting of early puberty in all cases. The difference was no longer significant at the end of the follow-up period, when endocrine changes were seen in 10/13 gliomas (p=0.62). The seven endocrine conditions which developed over time occurred in 3–9 years (5 cases of early puberty and one each of central hypothyroidism and central diabetes insipidus). All patients had a single endocrine disease, except for the only patient who had received radiotherapy, who had both central early puberty and hypothyroidism after 8 and 9 years respectively.
Presence of endocrine disease at diagnosis of sellar lesions, one year later, and at the end of the follow-up period in the complete cohort and by type of lesion.
| All | Gliomas | Germinal tumors | Craniopharyngiomas | Prolactinomas | Histiocytomas | Undiagnosed | |
| No. | 45 | 13 | 10 | 6 | 4 | 4 | 6 |
| At diagnosis | 24 | 3 | 10 | 3 | 2 | 4 | 1 |
| One | 11 | 3 | 1 | 0 | 2 | 3 | 1 |
| More than one | 13 | 0 | 9 | 3 | 0 | 1 | 0 |
| At one year | 30 | 3 | 10 | 6 | 4 | 4 | 1 |
| One | 12 | 3 | 1 | 0 | 3 | 3 | 1 |
| More than one | 18 | 0 | 9 | 6 | 1 | 1 | 0 |
| At the end of follow-up | 37 | 10 | 10 | 6 | 4 | 4 | 1 |
| One | 16 | 9 | 0 | 0 | 3 | 2 | 1 |
| More than one | 21 | 1 | 10 | 6 | 1 | 2 | 0 |
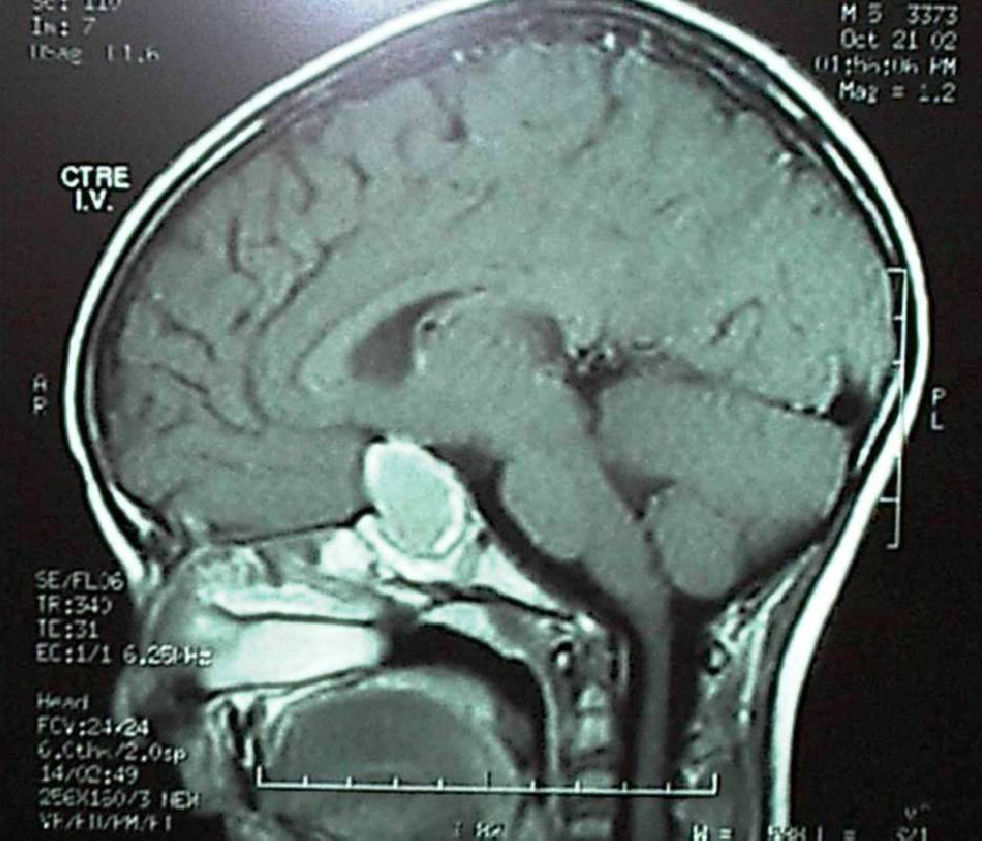
Complete anterior and posterior pituitary deficiency was found in 9 of 10 germinal tumors. All these lesions were greater than 20mm in size in neuroimaging at diagnosis. A single patient, who consulted for isolated diabetes insipidus and in whom was found pituitary stalk enlargement in neuroimaging, developed anterior deficiency three years later, consistent with tumor growth, before chemotherapy and radiotherapy were started. Panhypopituitarism (including central diabetes insipidus) was found at diagnosis in 3 of the 6 craniopharyngiomas. Panhypopituitarism occurred immediately after surgery in the other 3 cases (Fig. 2).
Discussion
Our data emphasize the importance of adequate identification of sellar lesions amenable to hormone treatment in children and adolescents and recognition of the symptoms and signs of endocrine disease to allow for early diagnosis and adequate management.
Although adenomas are rare in children, prolactinoma should mandatorily be ruled out because it responds to medical treatment. Prolactinoma mainly occurs in children older than 12 years, but has also been reported in prepubertal children. A simple prolactin measurement higher than 200ng/mL diagnoses the tumor. Thus, when any sellar or parasellar mass is found, even in prepubertal children and regardless of radiographic appearance, prolactin levels should be measured before considering surgery.2–5,8,9 Adenomas may be confused with craniopharyingiomas, which may also have a sellar origin. Although the former are usually solid and the latter cystic, long-standing adenomas may grow and have cystic areas due to tumor apoplexy (tumor necrosis or hemorrhage).10–12 In our series, while pubertal females were quickly diagnosed based on galactorrhea, males had no symptoms of hypogonadism (because pubertal delay is only considered to be pathological from the age of 14 years), consulted for headache, and had large macroadenomas with cystic areas at onset.
Dopamine agonists, mainly cabergoline, are the first choice for the treatment of prolactinoma because they significantly decrease tumor size in more than half the cases and normalize prolactin levels and restore gonadal function in more than three-quarters of the patients, preserving the remaining pituitary function. Surgery is therefore reserved for patients intolerant or resistant to medication.8,9,13,14 Pituitary hyperplasia or pseudoadenoma reactive to long-standing primary hypothyroidism is another example of a sellar mass that resolves with hormone treatment, as seen in one of our children.15
Chorionic gonadotropin is another hormone which should be tested before deciding lesion management. Almost half of the hypothalamic germinal tumors secrete chorionic gonadotropin, together with alpha-fetoprotein. Chemotherapy and radiotherapy are the treatment of choice for these tumors, and presence of these markers makes therefore biopsy unnecessary. As seen in our series, they usually cause hypopituitarism early, particularly central diabetes insipidus, often associated to anterior pituitary gland deficiencies. The few patients who have no panhypopituitarism at diagnosis experience it after oncological treatment.16–18
Craniopharyngioma is usually diagnosed based on neurological symptoms, although 80% of the patients have pituitary deficiencies which are not usually reasons for consultation. After surgery, virtually all the patients experience complete panhypopituitarism.1,2,5,19–22 As shown, most sellar lesions are associated at diagnosis to endocrine clinical symptoms, many of them long-standing. In a recently reported large series, two-thirds of the 176 patients reported endocrine symptoms within a mean of 6 months of the start with space-occupying symptoms.7 Exceptions include gliomas and granulomas from histiocytosis, where endocrine involvement is usually more sequential, and incidentalomas, which are clinically silent. It should be noted that many patients in our series had very long-standing (1–5 years) central diabetes insipidus and had not been referred for workup by their physicians, and in whom diabetes mellitus and renal or electrolyte disorders had only been ruled out.
Optic tract gliomas are the tumors diagnosed at an earliest age, particularly those (almost 50%) occurring in the setting of neurofibromatosis type 1. They are usually pilocytic astrocytomas of low-grade malignancy which often do not progress and even regress. They have usually no associated endocrine conditions at baseline, except in previously undiagnosed patients who attended the clinic for central early puberty or weight gain but no pituitary deficiencies. They may however develop over time, more probably if chemotherapy or radiotherapy has been administered, but also when no treatment has been administered.23–25 Excess growth due to GH hypersecretion has also been reported in these tumors.26 During follow-up of our children, hypopituitarism was related to prior primary treatment for the tumor, while early puberty also occurred in untreated patients.
One-fourth of the patients younger than 18 years with histiocytosis have pituitary deficiencies over the first 10 years. Almost all of them have diabetes insipidus, 50% GH deficiency and, to a lesser extent, other hormone deficiencies. Mean age at onset is 2.8 years for histiocytosis, 3.9 years for diabetes insipidus, and 7.7 years for GH deficiency. Multiple organ and craniofacial involvement is associated to hypothalamic involvement, and systemic treatment does not prevent them.27–29
Incidentalomas are found with increasing frequency in our patients. These are unsuspected masses discovered during workup for other unrelated diseases. Clinical practice guidelines for management of incidentaloma in adults are only available. Surgery is only needed for incidentalomas that cause neurological or visual signs and for those inducing excess secretion (except of prolactin). All other incidentalomas are monitored with MRI every 6 months.30
As shown, sellar area lesions require a multidisciplinary approach. Endocrine workup is indispensable, because endocrinologists may treat and resolve some of these lesions and their consequences (prolactinoma, reactive hyperplasia associated to hypothyroidism, other hormone deficiencies), or simplify diagnosis of other lesions (germinal tumors secreting chorionic gonadotropin). It is essential to recognize symptoms of endocrine disease to achieve earlier diagnosis, because many of these patients experience them at the start, quite earlier than neurological and visual symptoms. Endocrine monitoring should be maintained in all cases, because other hormone changes will occur over time.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: García-García E, González-Aguilera B, Gros N, Romero-Lluch A, Jiménez-Varo I, Martínez-Ortega AJ, et al. Diagnóstico y tratamiento endocrinológico de las lesiones del área selar en la edad pediátrica. Endocrinol Nutr. 2014;61:359–365.
