





Clostridioides difficile infection (CDI) has become a global healthcare challenge due to increases in its incidence and mortality rates. Faecal microbiota transfer (FMT) is postulated as a protocol to prevent CDI recurrence.
Material and methodsA donor faecal sample and patient faecal samples (pre-FMT and post-FMT) were analysed. The r16S gene was amplified and sequenced by NGS, and its diversity and taxonomy composition were examined.
ResultsMicrobial richness increased in post-FMT samples, and the β diversity studies grouped the samples into two clusters. One included the non-pathological samples (donor and pre-FMT samples), and the other included the pathological sample. The results obtained by Qiime2 and Bioconductor were similar.
ConclusionThe analysis showed an increase in taxonomic diversity after the FMT, which suggests its usefulness. Moreover, these results showed that standardisation of bioinformatics analysis is key.
La diarrea por Clostridioides difficile es un importante problema de salud pública, cuyo tratamiento es complejo. La transferencia de microbiota fecal (TMF) se postula como una terapia útil para prevenir recidivas.
Material y métodosSe analizaron seis muestras fecales, una procedente del donante y cinco del paciente antes y después de la TMF. Se amplificó y secuenció el gen 16Sr mediante secuenciación masiva y se estudió la diversidad y composición taxonómica.
ResultadosLa diversidad aumentó en las muestras post-TMF, y se identificaron dos clústeres, uno formado por las muestras no patológicas (donante y paciente post-TMF), y otro por la muestra patológica. Los resultados obtenidos a través Qiime2 y Bioconductor fueron similares.
ConclusiónEl análisis realizado demostró un incremento en la diversidad taxonómica del paciente tras la TMF, sugiriendo su utilidad. Además, los resultados obtenidos con Qiime2 y Bioconductor reflejaron la importancia de unificar los análisis bioinformáticos.
The incidence of Clostridioides difficile infection (CDI) has increased considerably over the last twenty years and thanks not only to the incidence, but also to its associated morbidity and mortality rates, CDI has become a global health problem. CDI is the main cause of nosocomial diarrhoea of infectious origin associated with the administration of antibiotics. It has led to an increase in in-hospital mortality, and generates significant healthcare costs.1
The treatment of choice for CDI is antibiotic therapy, which is effective in approximately 85% of cases. However, the incidence of recurrence is high, at around 30%, with the rate increasing to 45% after the first episode, and up to 75% in patients with multiple episodes of recurrence.2 A number of different factors have been associated with the increased risk of recurrence, including the need to remain on broad-spectrum antibiotic therapy, old age, or having suffered a previous episode of CDI.3 As all these factors affect the patient's own microbiota, there are theories that the underlying cause of relapses is dysbiosis due to colonisation by Clostridioides difficile (CD). Consequently, faecal microbiota transplantation (FMT) after the antibiotic treatment has been proposed as an effective method to prevent relapses in patients with this disorder.4
Various different computer-based tools are available to carry out microbiota analyses, such as Mothur,5 QIIME 26 and Bioconductor.7 Studies comparing the results obtained with Mothur and QIIME 2 have found no significant differences between them.8 However, as yet there are no published studies comparing these methods with the results obtained using Bioconductor.
Material and methodsSamples: the surplus of six stool samples were analysed, one from the recipient pre-transplant, considered pathological, and five non-pathological (from the donor, and from the recipient at 7, 30, 90 and 180 days after the procedure). The recipient was a patient who had already had six relapses of CDI. All the procedures were conducted following national ethical and legal standards, and in accordance with the guidelines established in the Declaration of Helsinki (2000).
FMT procedure: a faecal microbiota concentrate from a healthy donor was introduced by endoscopy. The donor was selected after screening which included a clinical questionnaire and microbiological studies of serum samples, faeces and nasal exudate. Presence of multiple pathogens was ruled out by culture and molecular techniques, as proposed in various clinical guidelines.9
Study of the microbiome by mass sequencing (next-generation sequencing [NGS]): the microbiota study followed the protocol recommended by Illumina.10 The QIIME 26 and Bioconductor7 programmes were used for the bioinformatic analysis.
The α diversity was studied by calculating the Shannon and Chao1 indices, and the β diversity through estimation of the unweighted Unifrac distance. Taxonomy was assigned using the SILVA database (Release 132).11 Differential taxa were identified through ANCOM-Composition and Gneiss in QIIME 2, DESeq2 and EdgeR in Bioconductor, and LefSe through the Galaxy server.
The comparative analysis of the results with QIIME 2 and Bioconductor was carried out by comparing the total taxa detection number (operational taxonomic units [OTU]) detected by both programmes, and the number of unidentified sequences. Student's t test was used for parametric variables and the Mann–Whitney U test for non-parametric variables.
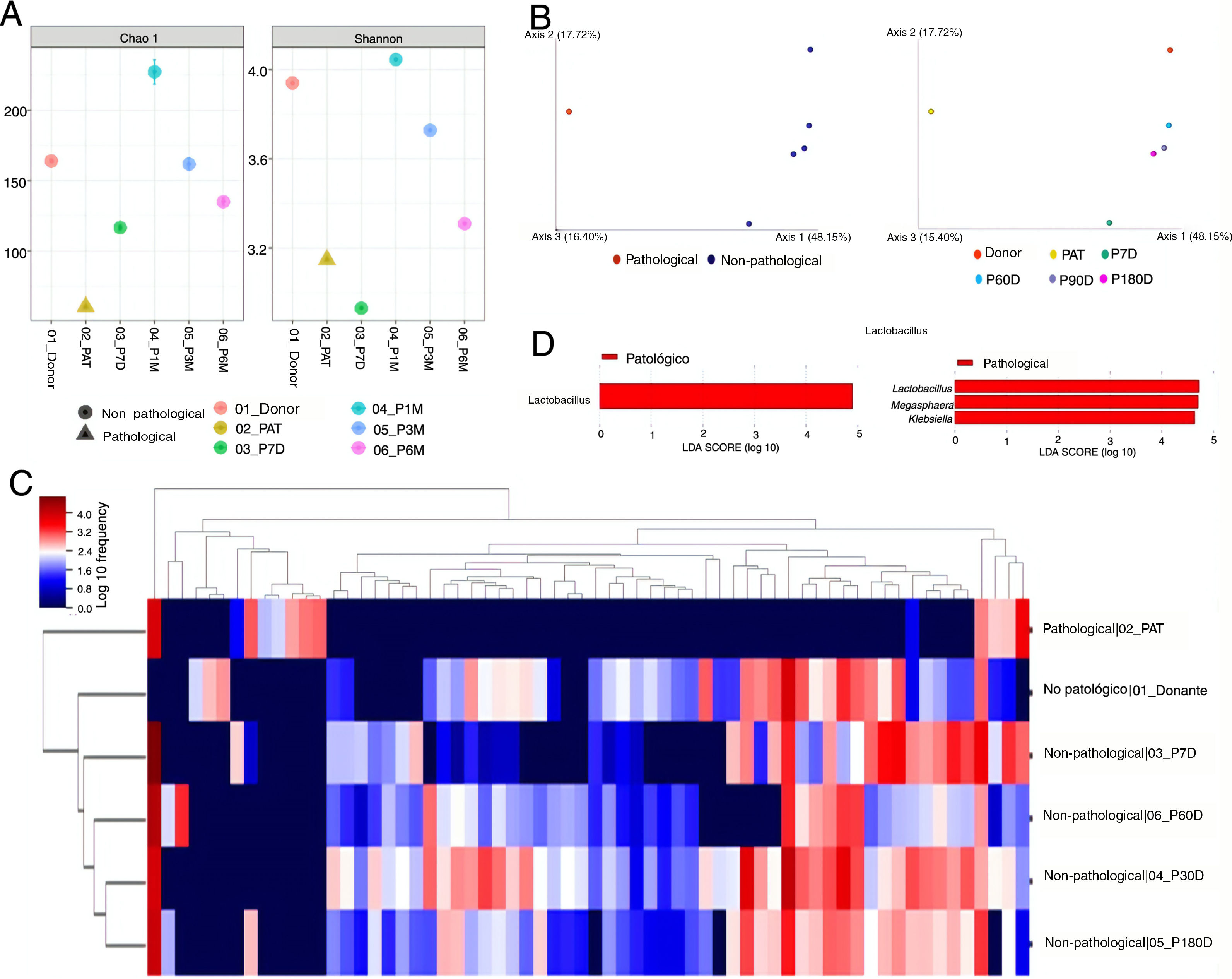
ResultsDiversity analysisThe lowest Chao1 and Shannon indices were those obtained for the pre-FMT samples, and at seven days post-FMT with both QIIME 2 and Bioconductor, so we could say these were the samples with the lowest α diversity. The sample obtained at 30 days post-FMT was the one with the greatest α diversity (Fig. 1A).
 α diversity analysis. Chao1 (left) and Shannon (right) indexes obtained with the Bioconductor programme. (B) β diversity analysis. Representation of the result of the calculation of the Unifrac unweighted distance, and the subsequent analysis of principal components with QIIME 2. (C) Heatmap obtained with QIIME 2. The sample data are horizontal. The information referring to the genera is vertical. (D) Differentially abundant genera using the LefSe algorithm. The image on the left corresponds to the one made with data from QIIME 2, and the one on the right to the one made with data from Bioconductor. Donor: sample obtained from the donor; PAT: sample obtained from the pre-FMT patient; P7D, P30D, P90D and P180D: samples obtained from the post-FMT patient at 7, 30, 90 and 180 days.")
(A) α diversity analysis. Chao1 (left) and Shannon (right) indexes obtained with the Bioconductor programme. (B) β diversity analysis. Representation of the result of the calculation of the Unifrac unweighted distance, and the subsequent analysis of principal components with QIIME 2. (C) Heatmap obtained with QIIME 2. The sample data are horizontal. The information referring to the genera is vertical. (D) Differentially abundant genera using the LefSe algorithm. The image on the left corresponds to the one made with data from QIIME 2, and the one on the right to the one made with data from Bioconductor. Donor: sample obtained from the donor; PAT: sample obtained from the pre-FMT patient; P7D, P30D, P90D and P180D: samples obtained from the post-FMT patient at 7, 30, 90 and 180 days.
The results for β diversity grouped the samples into two large clusters, separating the non-pathological samples (donor and post-FMT samples) from the pathological sample (pre-FMT). The samples with the most similarity were those of the donor, and of the patient at the longest times after the transplant, with these being far apart from the pre-FMT sample and the sample obtained seven days post-FMT (Fig. 1B).
Taxonomic composition analysisThe taxonomic composition analysis revealed that the non-pathological samples were more similar to each other, and different from the pre-FMT patient sample. Groups of microorganisms that characterised both pathological and non-pathological samples were observed (Fig. 1C).
The genera Lachnospira, Butyricimonas, Paraprevotella, Odoribacter and Anaerostipes were described as taxa associated with the non-pathological state by the DESeq algorithm. The ANCOM and Gneiss algorithms also identified some of these genera as associated with this state. Of the rest of the taxa identified as differentially abundant among the studied samples, seven were detected by at least three algorithms: Blautia, Bacteroides and Alistipes associated with the non-pathological samples; and Klebsiella, Veillonella, Megasphaera and Lactobacillus with the pathological state (Table 1).
Differentially abundant taxa found using the ANCOM, Gneiss, DESeq, EdgeR and LefSe algorithms. Those associated with the non-pathological state are marked in green and those associated with the pathological state in red.
| Taxa | ANCOM | Gneiss | DESeq | EdgeR | LEfSe |
|---|---|---|---|---|---|
| Blautia | x | x | x | ||
| Bacteroides | x | x | x | ||
| Alistipes | x | x | x | ||
| Klebsiella | x | x | x | x | x |
| Veillonella | x | x | x | x | |
| Megasphaera | x | x | x | x | x |
| Lactobacillus | x | x | x |
The results obtained in the diversity and taxonomy analysis with QIIME 2 and Bioconductor were similar. However, to check if there were differences in the OTUs, or in the taxonomic assignment, a comparison was carried out evaluating the difference between the total OTUs observed, and the number of sequences not identified by each method. The mean total OTUs observed in QIIME 2 was 140, and in Bioconductor this was 141 (p-value=0.97), so we can state that there were no significant differences.
The mean percentage of unidentified sequences for the QIIME 2 programme was 9.96%, and for Bioconductor this was 7.91%. In this case, the percentage was found to be slightly higher with the QIIME 2 programme, although a p-value=0.197 was obtained, so it was established that there were no differences in the taxonomic assignment.
Comparison of the algorithms used to identify the differentially abundant taxa revealed that the most restrictive was LefSe, which only detected one differential taxon (Lactobacillus) with the QIIME 2 data, and three (Lactobacillus, Megasphaera and Klebsiella) with the Bioconductor data (Fig. 1D). The least restrictive was DESeq, which identified a total of 34 genera associated with one or other state. The Gneiss algorithm was the one that detected a greater number of taxa which were also detected by other algorithms (7/11, 63.6%). In contrast, the DESeq algorithm was the one that indicated a greater number of taxa which did not coincide with other algorithms, a total of 28 out of 34 (82.3%) (Table 1).
DiscussionThe greatest risk factor in the development of CDI is the administration of antibiotics, which causes an alteration in the composition of the intestinal bacterial community, increasing the host's susceptibility to the pathogen.12 Regardless of the analysis system, our results showed that the patient's microbiota before the microbiota transplant process was different from that of the donor, considered as a healthy microbiota, both in its diversity and in its taxonomic composition.
Healthy microbiota contain trillions of bacteria, primarily Bacteroidetes, Firmicutes, Actinobacteria and Proteobacteria,13 and act as a natural barrier that prevents CDI. Changes in the bacterial composition of the microbiota after FMT include the appearance of different genera, such as Lachnospira, Butyricimonas, Paraprevotella, Odoribacter and Anaerostipes, which are not found in the pre-transplant patient's microbiome due to the dysbiosis generated by massive colonisation of the colon by CD.14 The same changes were also found in the samples analysed in this study where, over time, these genera, which were absent in the sample taken from the patient before the FMT, gradually started to be identified.
The results obtained with QIIME 2 and Bioconductor are similar, and both can therefore be used without waiting for variations in the detection of taxa associated with bioinformatic analysis. This type of study is essential to establish a common work flow in the analysis of these data, so that the results obtained are comparable regardless of where they are obtained. After using both programmes, QIIME 2 being a very powerful and complete tool, Bioconductor allowed for increased adaptability of the “pipelines” to different situations, as well as greater versatility, due to the existence of a multitude of packages. Therefore, although both options are equally valid, we might suggest the use of Bioconductor.
FundingThis study received no specific funding from public, private or non-profit organisations.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Ventero MP, Espinosa N, Jover R, Guillen Y, Merino E, Rodríguez JC. Evolución del microbioma intestinal en un proceso de transferencia de microbiota fecal (TMF) en un paciente con infección por Clostridioides difficile: análisis por NGS con diferentes programas bioinformáticos. Enferm Infecc Microbiol Clin. 2021;39:184–187.
