




Acromegaly is a clinical syndrome caused by the excessive production of growth hormone. It is associated with high morbidity and significantly increased mortality, mainly due to cardiovascular and respiratory complications, and cancer. Mortality is reduced to that of the general population following successful treatment, in other words, when insulin-like growth factor (IGF-I) and growth hormone values return to normal levels. Not all tumours associated with this syndrome benefit from cost-effective early diagnosis programmes. An in-depth knowledge on the part of clinicians of the morbidity and mortality associated with acromegaly, allowing them in many cases to anticipate the expected clinical course of the disease, is the best therapeutic and follow-up strategy in these patients.
La acromegalia es un síndrome clínico producido por la secreción excesiva de hormona del crecimiento. Conlleva una gran morbilidad y un aumento significativo de la mortalidad, principalmente por complicaciones cardiovasculares, respiratorias, así como un aumento en la prevalencia del cáncer. La mortalidad se equipara a la de la población general cuando se consigue la curación de la enfermedad, esto es, la normalización analítica de los valores de IGF-I (factor de crecimiento similar a la insulina tipo I) y hormona del crecimiento. No todos los tumores asociados a esta entidad son subsidiarios de programas coste-efectivos para su diagnóstico temprano. La mejor estrategia terapéutica y de seguimiento en estos pacientes es el conocimiento por el médico responsable de la morbimortalidad asociada a esta entidad, adelantándonos en muchos de los casos al curso evolutivo esperable.
Acromegaly is a disease caused by the chronic and inappropriate secretion of excess growth hormone (GH), which starts after growth plates have closed.
When the increased secretion happens whilst these are still open, it results in gigantism.
Although knowledge of the disease is very old, it was Pierre Marie who coined the term “acromegaly” in 1886, which comes from the Greek acros (end) and megas (big).
Without treatment, most patients with this disease have a life expectancy of around 60 years,1–3 with mortality being double or triple of that expected,4 mainly due to metabolic, respiratory, cardiovascular, and cerebrovascular disorders associated with it.5–8 The risk of neoplasms is also increased in many organs, such as in the digestive tract, lungs, breast, prostate, kidney and brain.10–17 Benign tumours are more common than malignant ones.14,18
EpidemiologyAcromegaly is a rare disease with an incidence of 3–6 new cases per million people and per year.4,8 It is equally common in both sexes and the age at diagnosis is 40 years in men and 45 years in women.
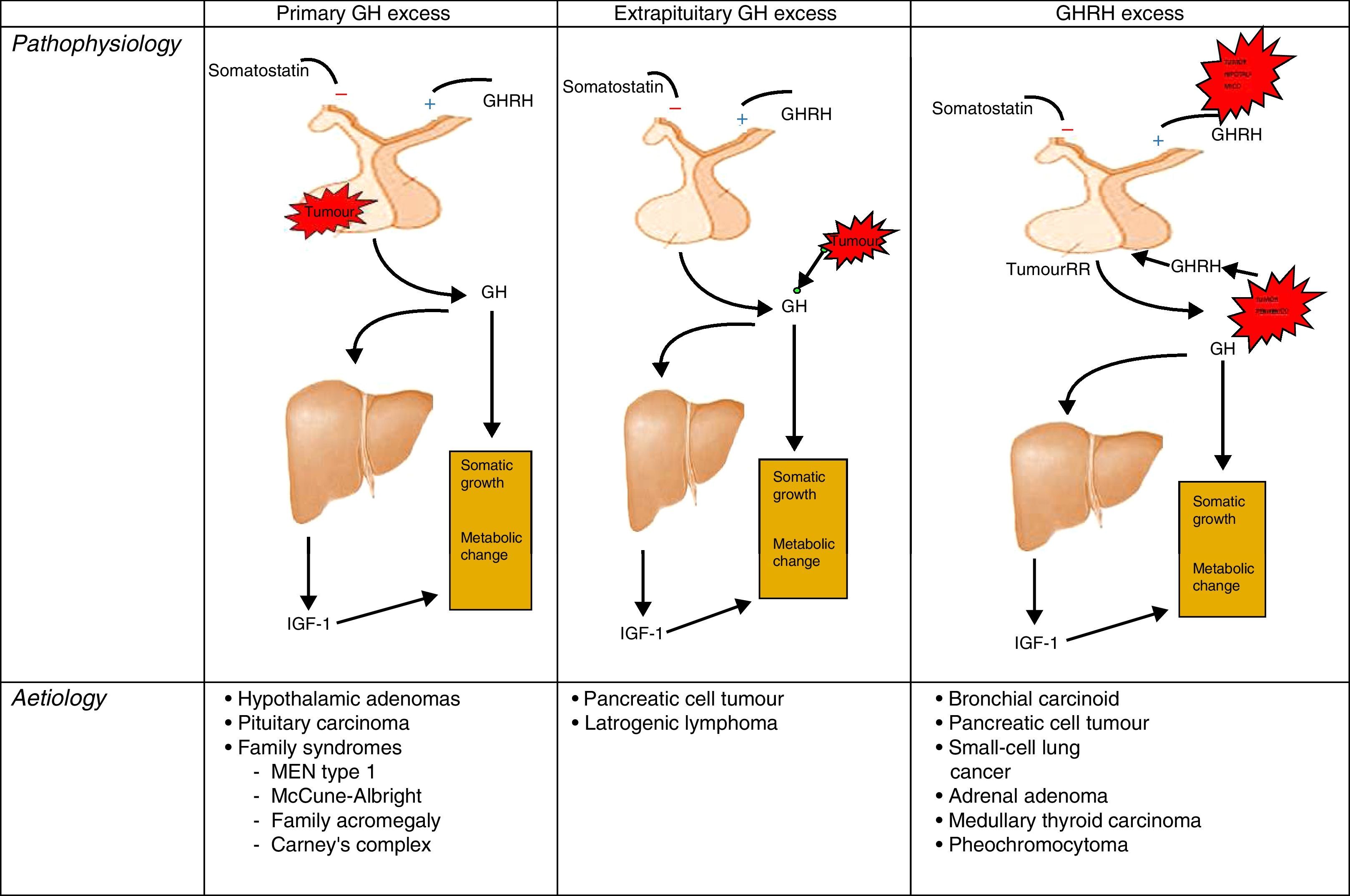
AetiopathogenesisThe cause of acromegaly syndrome is chronic and prolonged production of excess GH. More than 99% of acromegaly cases are due to a GH-secreting pituitary tumour.19,20
The remaining percentage corresponds to the extrahypothalamic-pituitary gland secretion of GH, generally by neuroendocrine tumours or by ectopic secretion of the GH releasing hormone (GHRH). These GHRH-producing tumours are called GHRHoma, and most cases originate in the lungs (47–53%) or pancreas (29–30%), and more rarely in the small intestine (8–10%).21–23
The GH-secreting pituitary adenomas are relatively common tumours, accounting for 30% of pituitary tumours, which may exclusively produce GH, or mixed adenomas that segregate GH and prolactin, but these last ones are rarer. 75% of cases are macroadenomas and tumours over 10mm are the bulkiest with the most aggressive behaviour amongst younger patients.
GH-producing pituitary carcinomas are very rare and diagnosed by the presence of metastasis in meninges, liver, bones, or lymph nodes.23
PathophysiologyRecall that the release of GH in adults is very limited and maintains its predominantly nocturnal pulsating character during periods of growth. In acromegaly, the 24-h GH secretion increases (by more than 2μg/L), conserving the pulsating character and the nocturnal peak. GH inhibition does not occur and there is the occasional appearance of a paradoxical elevation in response to the glucose24 (Fig. 1).

Different origins of GH oversecretion. Taken and modified from Rúa et al.21
Dopaminergic agents, including bromocriptine, boost GH in healthy individuals, whilst in acromegaly patients, they inhibit it.
GH oversecretion results in a similar growth factor elevation to insulin-like growth factor 1 (IGF-1) and the anti-insulin action of the GH, exacerbates pre-existing diabetes mellitus or promotes its onset.
DiagnosisFor biochemical diagnosis, random assessments of GH must not be used because there are multiple clinical situations other than acromegaly that can occur with abnormal concentrations of GH: taking oestrogen, hypoglycaemia, kidney failure, etc.21
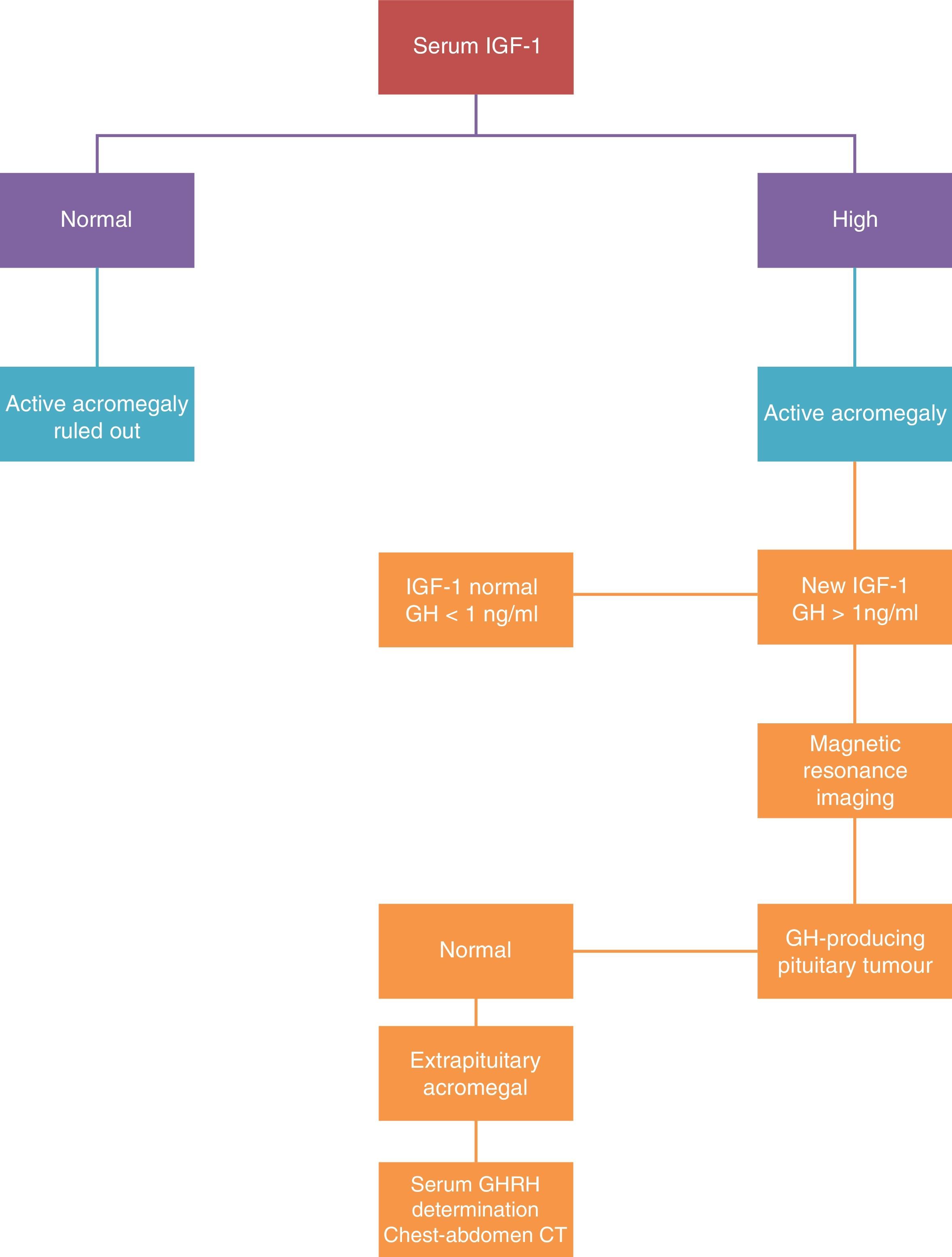
The diagnosis is based on demonstrating excessive GH production for 24h, or the changes in the excretion dynamics.
There are two tests for selective assessment: IGF-I assessment and suppressing GH secretion with oral glucose tolerance testing. The disease diagnosis is confirmed when the IGF-1 levels are confirmed as being above the normal values for age and sex, and when the GH assessment after the oral glucose tolerance test is greater than 2μg/L (Fig. 2).

The measured IGF-1 concentration is correlated with the disease activity, and this assessment is the one used in clinical practice to monitor the disease. The treatment is considered as curative when normal levels of IGF-1 are reached and the GH is reduced to under 2μg/L after the oral glucose tolerance test. When these levels are achieved, morbimortality is equal to that of the general population.
Plasma GHRH assessment is only of interest if an ectopic source is suspected.
An MRI scan is the morphological technique of choice for studying pituitary tumours. In agreement with neuroradiological findings, the tumours are classified as localised (stage I microadenoma and stage II macroadenoma) or invasive (stage III and IV).
If ectopic GHRH secretion is suspected, a thoracoabdominal CAT scan, digestive study, endoscopic ultrasound, and arteriography must be performed.25
Both symptoms and morbidity of the acromegaly are due to the mass effect of the pituitary tumour and the GH oversecretion.
HistoryThe symptoms usually appear insidiously, with changes that often go unnoticed. Therefore, more than 10 years can pass from the beginning of the disease until diagnosis.26
Headaches are more common than in any other type of pituitary adenomas. In the case of invasive tumours, hemianopia and even rhinorrhoea can occur.
Given that the growth plates are closed when the GH oversecretion begins, there is no possibility of longitudinal growth, which causes an initial increase of the soft tissues and then an increase of the extreme parts of the skeleton. The nose, lips, ears, tongue, hands, feet, jaw, and supraorbital ridge and zygomatic arch experience progressive growth, resulting in a characteristic facial appearance. The laryngeal growth grants a “deep voice”. Skin thickness increases, there is hyperhidrosis and hyperpigmented or molluscum-like skin lesions often occur (skin tags). Carpal tunnel syndrome occurs in 30–58% of these patients. After effective treatment, the soft tissues regress, but bone deformities persist.
HTN is three to four times more common than in the general population, affecting one third of acromegaly patients. This is due to GH promoting sodium retention as a direct effect on the renal pump. Around one third of patients show heart involvement due to left ventricular concentric hypertrophy and asymmetric septal hypertrophy.27 This involvement is related to the disease progression time. Despite everything, only 15–20% of the patients show cardiac symptoms in the form of coronary heart disease, heart failure, or arrhythmias.28
Sixty percent of patients present sleep apnoea, mainly due to upper airway obstruction, although it could also have a central origin.12
With regard to associated endocrine disease, 3–7% of these patients generally present non-toxic multinodular goitre and hyperthyroidism is uncommon.29
Between 25% and 60% of acromegalies present alterations in the hydrocarbon metabolism in the form of insulin resistance, but only 10–12% of those patients will develop clinical diabetes mellitus.30
Anterior pituitary failure from tumour compression is rare. The PRL secretion is primarily affected with a hyperprolactinemia that can occasionally coincide with galactorrhoea.31
Acromegaly can make up part of an MEN type 1, and is almost always associated with hyperparathyroidism and less commonly with a pancreatic tumour. MEN type 1 is suspected as the cause of acromegaly if associated with hypercalcaemia.
Regarding effects on the abdominal organs, they can lead hepatosplenomegaly that may or may not coincide with changes in laboratory values. Mild hypertransaminasaemia is the most common change and associated portal hypertension is not described in these cases. The incidence of gallbladder lithiasis is also rising.32
Macroglossia, dolichocolon, and colon diverticula are more common amongst these patients.33
Acromegaly and neoplasmsThe increase of serum and tissue GH and IGF-1 is related to a greater mitogenesis and cell proliferation,24 which increases the incidence of both benign and malignant neoplasms.14 There are in vitro, in vivo, and epidemiological studies linking excess GH with the proliferation of intestinal cells.34–38 This tumour risk is equal to that of the general population when the disease is controlled with treatment.16
The prevalence of gastrointestinal tumours is increased in most organs, which makes colon polyps and colorectal cancer (CRC) the oldest described association and also the best documented.
The description of this incidence has varied over the years, from the first retrospective epidemiological studies39–41 to the more modern controlled studies,42–45 and it is increasing in all cases. A recent meta-analysis from 200846 that looked at the risk of colorectal cancer in acromegaly patients shows that there is increased risk, both with regards to adenomatous polyps and hyperplastic polyps as well as with colorectal cancer. Of the controlled studies in the meta-analysis, perhaps the most significant one is the multicentre study conducted in Italy by Terzolo et al.43 In a cohort of 235 acromegaly patients alongside 233 control patients, a systematic colonoscopy was carried out in both groups. In the acromegaly cohort, a total of 65 colonoscopies were detected with pathological findings (27.7%), i.e. hyperplastic polyps, adenomatous polyps, or CRC, versus 36 in the control cohort (15.5%). Adenomas were found in 23.4% of the acromegaly patients versus 14.6% of the control group, and 4.3% of the acromegaly patients had CRC versus 0.9% of the control patients. This ratio of prevalence in colorectal cancer for control patients is in agreement with the 0.6–1.0% published in a series of CRC colonoscopy screenings.47,48
In this same study by Terzolo et al., after stratifying by age, it was determined that within the pathological findings of the colonoscopies for both hyperplastic and adenomatous polyps and CRC amongst acromegaly patients under 40 years old, the percentage of pathological examinations was significantly greater (19.3%) than in the control group with the same age range (4.4%). This risk difference is lower in those over 50 years old where for acromegaly patients it is 31.1% and for the control group it stands at 20%. Upon stratifying the findings by lesion type, 19.1% of the patients with acromegaly presented hyperplastic polyps in comparison with 9.4% of the control group patients. Adenomatous polyps were found in 23.4% of the acromegaly patients versus 14.6% from the control cohort. Therefore, this study concludes that acromegaly presents a moderate but definitive risk increase for colon neoplasms and that they happen at an earlier age than within the general population.
The cumulative lifelong CRC rate in our field is around 5.6%.49
Colon polyps in acromegaly are more commonly located in the ascending colon and are histologically more advanced at the time of diagnosis.50 However, other studies suggest that the incidence of colon adenomas is similar to that of the general population and that the incidence of hyperplastic polyps is higher.42
It is recommended to monitor the occurrence of CRC in all acromegaly patients over 40 years old who have had the illness for over 10 years and/or a family history of colon cancer.51
A colonoscopy is the test of choice for this screening given that it has proven to be superior to faecal occult blood test.52
Regarding the colorectal adenoma screening and monitoring strategy, the 2010 British guidelines include a specific section for screening and monitoring acromegaly patients, within the at-risk populations.53 There is little scientific evidence in this aspect and it is therefore better to give recommendations based on clinical guidelines because they gather scientific evidence better than specific studies.
According to these guidelines, a complete, quality, baseline colonoscopy must be carried out in acromegaly patients at age 40 with endoscopic follow-ups every 5 years if the endoscopies are normal.54
It is more difficult to obtain a good quality colonoscopy for the general population due to the fact that dolichocolon is common and it has a higher transit time. Therefore, many authors recommend doubling the evacuant solution dosage.55
When an adenomatous polyp has been detected in the first colonoscopy, it is recommended to perform rigorous follow-ups by way of endoscopic examinations every two or three years, especially if the polyp is bigger than 10mm and mainly in patients who show an insufficient response to medical treatment.56 Although, according to the 2010 British guidelines, the recommendations to monitor the polyp are the most typical.
Elevated GH levels and the presence of polyps in the baseline colonoscopy are factors related to polypoid relapse.56
Of the rest of the tumours, no higher incidence has been described versus the general population. However, there is a wide spectrum of malignant tumours described in the literature in acromegaly patients.17,57–59
It is rare, but there are cases of patients with acromegaly and multiple tumours.60 So much so that it was at the root of a clinical case that we are managing with acromegaly, schwannoma, classic seminoma, colon adenomatous polyps and gastric adenocarcinoma, that we, the authors, decided to update knowledge on the topic.
The standardised incidence rate for gastrointestinal neoplasms at other levels is also higher, but to a lesser extent. The widest corresponding series, in a cohort of 1041 acromegaly patients,9 identifies an oesophageal cancer rate of 3.1 versus the general population, with a gastric and small intestine cancer rates of 2.5 and 6.2, respectively.
In the context of gastric cancer, IGF-1 overexpression is described in the tumour tissue of the surgical specimen.61 This fact, whether or not it is associated with Helicobacter pylori infection, could establish the occurrence of these types of tumours at an earlier age with a more aggressive histology. This was so in the clinical case that we are managing.
Different studies suggest a greater incidence of lung, breast, thyroid and prostate tumours,10–17 although the latter is more controversial and there are studies that refer to a similar risk to that of the general population.15
PrognosisThis disease shortens life expectancy and doubles or triples the expected death rate.6
Deaths primarily occur due to cardiovascular causes in patients under 60 years old.
Respiratory causes account for 25% of the deaths and the third most common cause of death is tumours.
The disease is also associated with a significant increase in morbidity: HTN, DM, arthrosis and myocardiopathy (specific or non-specific), are more common than in the general population.30
TreatmentThe mainstays of treatment would be the prevention of neoplasm pathologies, especially those of colorectal origin, and respiratory and cardiovascular disorders.25
The treatment objectives are to eradicate the tumour and normalise GH and IGF-1 secretion. This must all be done to try to preserve thyroid function.
Transsphenoidal surgery is indicated for all pituitary adenomas except for those that show wide suprasellar extension. In experienced hands, it is possible to reach a cure with strict criteria in 45–45% of the cases.62,63
The prognostic factors that influence curative resection are the size of the tumour, its invasiveness, and the pre-surgical GH concentrations.64
Fractionated stereotactic radiotherapy is recommended when it has been impossible to normalise the GH after transsphenoidal surgery and very rarely as primary treatment.65
The percent GH decline versus its baseline concentration varies between 42% and 60% after two years and 75–80% after 5 years. The slow decline manages to control the illness in 70% of the cases after 10 years.65,66
Medical treatmentSomatostatin analogues are currently the most commonly used drugs.
They achieve GH figures below 2.5μg/L in 40–50% of cases and they normalise the IGF-1 in 40% of cases.67
They also prompt a moderate reduction of the tumour size. They are the only effective drugs for acromegaly caused by ectopic GHRH secretion and are prescribed when other therapies have failed and for preoperative treatment.68,69
Dopamine agonists have a low effectiveness, managing to lower the GH<5μg/L in only 20% of cases and normalise the IGF-1 in 10%. In any case, they improve symptoms in up to 60% of the cases.70
The GH-receptor blockers are not systematically used.
ConclusionThe treatment objective to decrease morbimortality from acromegaly is to cure, or failing this, control the disease.
The tumour pathology has a variable incidence that is higher than that of the general population. With regards to CRC, screening colonoscopies are recommended in acromegaly patients starting the age of 40, who have had the disease for more than 10 years, or have a family history of CRC.
Since gastrointestinal cancer also presents a discreet increase of the relative risk in comparison with the general population, we the authors, propose the joint performance of a gastroscopy and a colonoscopy, at least in the baseline study. The cost-benefit ratio of this screening needs wider study.
Finally, the greater awareness of morbimortality associated with this entity is the best therapeutic and monitoring strategy, which allows us to anticipate progression in many cases.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Calderón MR, Delgado E, García Campos F. Acromegalia y tumores asociados: ¿qué debemos saber los gastroenterólogos? Gastroenterol Hepatol. 2017;40:41–47.
