




La acromegalia es un síndrome clínico producido por la secreción excesiva de hormona del crecimiento. Conlleva una gran morbilidad y un aumento significativo de la mortalidad, principalmente por complicaciones cardiovasculares, respiratorias, así como un aumento en la prevalencia del cáncer. La mortalidad se equipara a la de la población general cuando se consigue la curación de la enfermedad, esto es, la normalización analítica de los valores de IGF-I (factor de crecimiento similar a la insulina tipo I) y hormona del crecimiento. No todos los tumores asociados a esta entidad son subsidiarios de programas coste-efectivos para su diagnóstico temprano. La mejor estrategia terapéutica y de seguimiento en estos pacientes es el conocimiento por el médico responsable de la morbimortalidad asociada a esta entidad, adelantándonos en muchos de los casos al curso evolutivo esperable.
Acromegaly is a clinical syndrome caused by the excessive production of growth hormone. It is associated with high morbidity and significantly increased mortality, mainly due to cardiovascular and respiratory complications, and cancer. Mortality is reduced to that of the general population following successful treatment, in other words, when insulin-like growth factor (IGF-I) and growth hormone values return to normal levels. Not all tumours associated with this syndrome benefit from cost-effective early diagnosis programmes. An in-depth knowledge on the part of clinicians of the morbidity and mortality associated with acromegaly, allowing them in many cases to anticipate the expected clinical course of the disease, is the best therapeutic and follow-up strategy in these patients.
La acromegalia es una enfermedad producida por la hipersecreción crónica e inapropiada de hormona del crecimiento (GH), que se inicia después del cierre de los cartílagos de conjunción.
Cuando el aumento de secreción acontece mientras estos permanecen abiertos se origina el gigantismo.
Aunque el conocimiento de la enfermedad es muy antiguo, fue Pierre Marie quien en 1886 acuñó el término «acromegalia», que deriva del griego acros (extremo) y megas (grande).
Sin tratamiento, la mayoría de los pacientes con esta enfermedad tienen una esperanza de vida entorno a los 60 años1–3, siendo la mortalidad el doble o el triple de la esperada4, principalmente por los trastornos metabólicos, respiratorios, cardiovasculares y cerebrovasculares que asocia5–8. Está aumentado además, el riesgo de neoplasias en multitud de órganos, como son el tracto digestivo, el pulmón, mama, próstata, riñón y cerebro10–17. Los tumores benignos son más frecuentes que los malignos14,18.
EpidemiologíaLa acromegalia, es una enfermedad rara, con una incidencia de 3 a 6 nuevos casos por millón de habitantes y año4,8. Presenta igual frecuencia en ambos sexos y la edad al diagnóstico es de 40 años en el hombre y 45 en la mujer.
EtiopatogeniaLa causa del síndrome acromegálico es la producción excesiva de GH de forma crónica y prolongada. Mas del 99% de los casos de acromegalia se deben a un tumor hipofisario secretor de GH19,20.
El porcentaje restante corresponde a la secreción extrahipotalámica-hipofisiaria de GH, por tumores generalmente neuroendocrinos, o por la secreción ectópica del factor estimulador de GH (GHRH). Estos tumores productores de GHRH son los denominados GRFnoma, siendo en la mayoría de los casos de origen pulmonar (47-53%) o pancreático (29-30%), y menos frecuentemente de origen en el intestino delgado (8-10%)21–23.
Los adenomas hipofisarios secretores de GH son tumoraciones relativamente frecuentes, representando el 30% de los tumores de la hipófisis, pudiendo ser productores de GH exclusivamente, o adenomas mixtos, que segregan GH y prolactina, siendo estos últimos menos frecuentes. El 75% de los casos se trata de macroadenomas, tumores mayores de 10mm siendo el tamaño más voluminoso y el comportamiento más agresivo en los pacientes más jóvenes.
Los carcinomas hipofisarios productores de GH son muy raros diagnosticándose por la presencia de metástasis en meninges, hígado, huesos o ganglios linfáticos23.
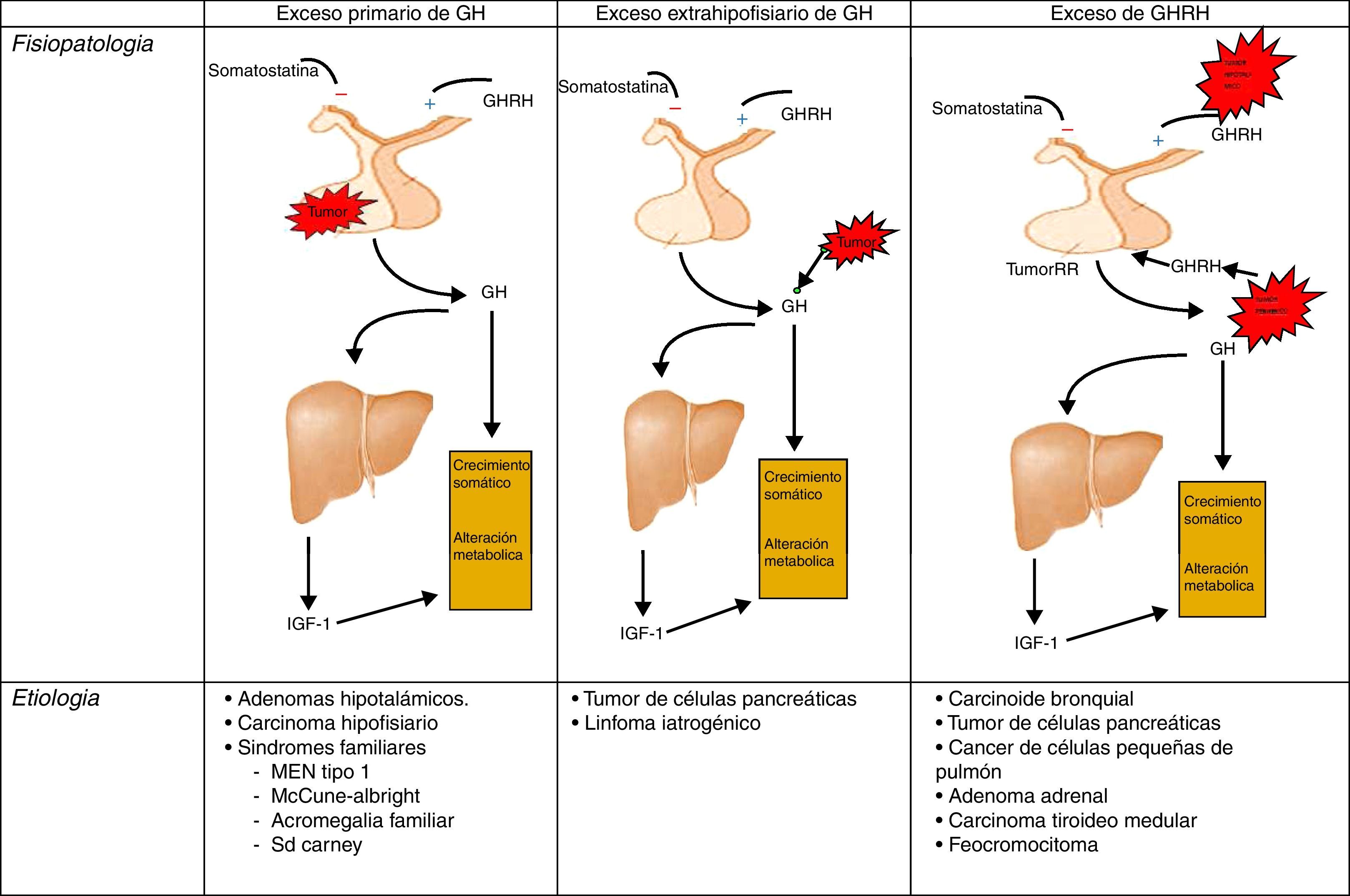
FisiopatologíaRecordemos, que la liberación de GH en el adulto es muy escasa, y mantiene el carácter pulsátil predominantemente nocturno de los periodos de crecimiento. En la acromegalia, la secreción de GH en 24 h está aumentada (por encima de 2μg/l), conservándose el carácter pulsátil y el pico nocturno, no produciéndose la inhibición de GH y apareciendo, en algún caso, elevación paradójica en respuesta a la glucosa24 (fig. 1).

Diferentes orígenes de la hipersecreción de GH. Tomada y modificada de Rúa et al.21.
Los agentes dopaminérgicos, entre ellos la bromocriptina, en individuos sanos estimulan la GH, mientras que en el paciente acromegálico, la inhiben.
La hipersecreción de GH ocasiona la elevación del factor de crecimiento similar a la insulina tipo I (IGF-I) y la acción antiinsulínica de la GH, agrava la diabetes mellitus preexistente o favorece su aparición.
DiagnósticoPara el diagnóstico bioquímico, las determinaciones aleatorias de GH no deben utilizarse, pues existen múltiples situaciones clínicas distintas a la acromegalia que pueden cursar con concentraciones anormales de GH: toma de estrógenos, hipoglucemia, insuficiencia renal, etc.21.
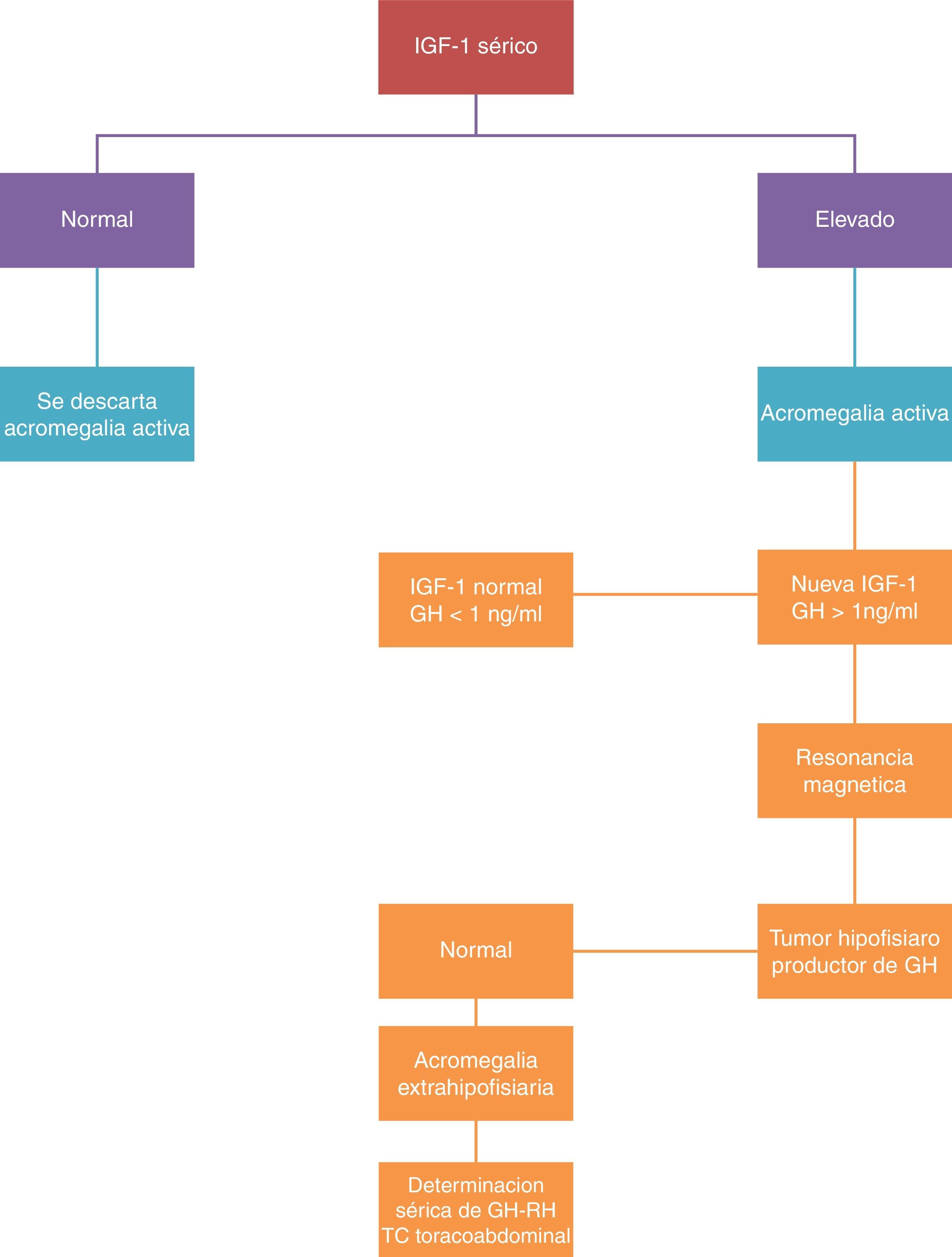
El diagnóstico se basa en la demostración de la excesiva producción de GH durante las 24 h, o de las alteraciones en la dinámica de excreción.
Se dispone de dos pruebas para la determinación selectiva: la determinación de IGF-I y la supresión de lamsecreción de GH con la sobrecarga oral de glucosa. Se confirma el diagnóstico de la enfermedad cuando se confirman niveles de IGF-I por encima de los valores normales para edad y sexo, y cuando la determinación de GH tras sobrecarga oral de glucosa es mayor de 2μg/l (fig. 2).

La medida de concentración de IGF-I se correlaciona con la actividad de la enfermedad, siendo esta determinación la utilizada en la práctica clínica para monitorización de la enfermedad. Se considera el tratamiento como curativo, cuando se alcanzan niveles normales de IGF-I y la GH se reduce por debajo de 2μg/l tras la sobrecarga oral de glucosa. Cuando se consiguen estos niveles, la morbimortalidad se iguala a la de la población general.
La determinación de GHRH en plasma tiene interés solo si se sospecha una fuente ectópica del mismo.
La RM es la técnica morfológica de elección en el estudio de tumores hipofisarios. De acuerdo con los hallazgos neurorradiológicos se realiza la clasificación de los tumores como localizados (estadio I microadenoma y estadio II macroadenoma) o invasores (estadio III y IV).
Ante la sospecha de una secreción ectópica de GHRH, debe realizarse una TAC toracoabdominal, estudio digestivo, ecoendoscopia y arteriografía25.
Tanto la clínica como la morbilidad de la acromegalia, se deben al efecto masa del tumor hipofisario y a la hipersecreción de la GH.
ClínicaLos síntomas suelen presentarse de forma insidiosa, con cambios que frecuentemente pasan desapercibidos por lo que pueden trascurrir más de 10 años desde el inicio de la enfermedad hasta el diagnóstico26.
La cefalea, es más frecuente que en otro tipo de adenomas hipofisarios. En caso de tumores invasivos, pueden aparecer hemianopsia e incluso rinorrea.
Al encontrarse cerrados los cartílagos de conjunción cuando comienza la hipersecreción de GH, no existe la posibilidad de crecimiento longitudinal, originándose un aumento de los tejidos blandos inicialmente y de las porciones extremas del esqueleto posteriormente. Nariz, labios, orejas, lengua, manos, pies, mandíbulas y arcos supraciliares y cigomáticos experimentan un crecimiento progresivo, dando lugar al aspecto facial característico. El crecimiento laríngeo confiere «voz cavernomatosa». El grosor de la piel aumenta, existe hipersudoración y frecuentemente aparecen lesiones cutáneas hiperpigmentadas o moluscoides (skin tags). El síndrome del túnel carpiano aparece en el 30-58% de estos pacientes. Tras el tratamiento efectivo, se produce regresión de los tejidos blandos, persistiendo las deformidades óseas.
La HTA es de tres a cuatro veces más frecuente que en la población general, padeciéndola un tercio de los pacientes acromegálicos y debiéndose a que la GH favorece la retención de sodio por efecto directo sobre la bomba renal. Alrededor de un tercio de los pacientes presenta afectación cardíaca por hipertrofia concéntrica del ventrículo izquierdo e hipertrofia septal asimétrica27. Esta afectación se relaciona con el tiempo de evolución de la enfermedad. A pesar de todo, solo el 15-20% de los enfermos presenta sintomatología cardíaca en forma de enfermedad coronaria, insuficiencia cardíaca o arritmias28.
El 60% de los pacientes presentan apnea del sueño, fundamentalmente por obstrucción de las vías aéreas superiores, aunque puede tener también un origen central12.
En relación a la afección endocrina asociada, un 3-7% de estos pacientes presenta bocio multinodular, en general no tóxico siendo el hipertiroidismo, infrecuente29.
Entre el 25 y el 60% de las acromegalias presentan alteraciones del metabolismo hidrocarbonatado en forma de resistencia a la insulina, pero solo un 10-20% de estos pacientes desarrollará diabetes mellitus clínica30.
La insuficiencia anterohipofisiaria por compresión tumoral es rara. Se afecta en primer lugar la secreción de PRL, con una hiperprolactinemia que ocasionalmente puede cursar con galactorrea31.
La acromegalia puede formar parte de un MEN tipo 1, casi siempre asociado con hiperparatiroidismo y con menor frecuencia a un tumor pancreático. Sospecharemos un MEN tipo 1 como causa de una acromegalia cuando asocie hipercalcemia.
En cuanto a la afectación visceral abdominal, puede condicionar hepatoesplenomegalia que puede cursar con cambios analíticos o no; la hipertransaminasemia leve es la alteración más frecuente, y no está descrita la hipertensión portal asociada en estos casos. Se encuentra también aumentada la incidencia de litiasis vesicular32.
Son más frecuentes en estos pacientes la macroglosia, el dolicocolon y los divertículos colónicos33.
Acromegalia y neoplasiasEl aumento sérico y en los tejidos de GH e IGF-1, se relaciona con una mayor mitogénesis y proliferación celular24 estando aumentada la incidencia de neoplasias, tanto benignas como malignas14. Existen estudios in vitro, in vivo y epidemiológicos que asocian el exceso de GH con la proliferación de células intestinales34–38. Este riesgo tumoral se equipara al de la población general cuando con tratamiento se consigue control de la enfermedad16.
La prevalencia de los tumores gastrointestinales está aumentada en la mayoría de los órganos, siendo la asociación más antiguamente descrita y la mejor documentada la de los pólipos colónicos y el cáncer colorrectal (CCR).
La descripción de esta incidencia ha variado a lo largo de los años, desde los primeros estudios epidemiológicos retrospectivos39–41 hasta los estudios controlados más modernos42–45, estando aumentada en todos los casos. Un reciente metaanálisis del año 200846 que analiza el riesgo de cáncer colorrectal en pacientes acromegálicos, muestra que está aumentado el riesgo, tanto de pólipos adenomatosos como hiperplásicos, así como del cáncer colorrectal. De los estudios controlados, sobre los que se realiza el metaanálisis, quizás el más significativo es el estudio multicéntrico realizado en Italia por el grupo de Terzolo et al.43. Sobre una cohorte de 235 pacientes con acromegalia frente a 233 controles, se realizó sistemáticamente colonoscopias en ambos grupos. En la cohorte de acromegálicos, se detectaron un total de 65 colonoscopias con hallazgos patológicos (27,7%), es decir, pólipos hiperplásicos, adenomatosos o CCR, frente a 36 en la cohorte de controles (15,5%). Se encontraron adenomas en el 23,4% de los pacientes acromegálicos frente al 14,6% de los controles y 4,3% de CCR en acromegálicos frente al 0,9% de los pacientes controles. Este ratio de prevalencia en cáncer colorrectal de pacientes controles es concordante con el 0,6-1,0% publicado en series de cribado de CCR de colonoscopias47,48.
En este mismo estudio de Terzolo et al., al estratificar por edad, los hallazgos patológicos de las colonoscopias, tanto pólipos hiperplásicos, como adenomatosos y CCR, se determinó que en pacientes acromegálicos menores de 40 años, el porcentaje de exploraciones patológicas era significativamente mayor (19,3%) que en la población control del mismo rango de edad (4,4%). Esta diferencia de riesgo es menor en los mayores de 50 años, donde en acromegálicos es del 31,1%, frente al 20% de los controles. Al estratificar los hallazgos por tipo de lesiones, el 19,1% de los pacientes con acromegalia presentaban pólipos hiperplásicos, frente al 9,4% de los pacientes controles. Un 23,4% de los pacientes acromegálicos presentaban pólipos adenomatosos, frente al 14,6% de los controles. Por tanto, este estudio concluye que la acromegalia presenta un moderado pero definitivo incremento del riesgo de neoplasias colónicas y que, estas acontecen a más temprana edad que la población general.
La tasa acumulada de CCR a lo largo de la vida, en nuestro medio es de en torno al 5-6%49.
Los pólipos colónicos en la acromegalia, se localizan más frecuentemente en colon derecho y son histológicamente más avanzadas en el momento del diagnóstico50. Sin embargo, otros estudios sugieren que la incidencia de adenomas colónicos es similar a la de la población general y que está aumentada la incidencia de pólipos hiperplásicos42.
Se recomienda vigilar la aparición de CCR en todos los pacientes acromegálicos mayores de 40 años, en los que tienen enfermedad de más de 10 años de evolución y/o historia familiar de cáncer de colon51.
La colonoscopia es la prueba de elección para la realización de este cribado, dado que ha demostrado ser superior a la realización del test de sangre oculta en heces52.
En cuanto a la estrategia de cribado y vigilancia de adenomas colorrectales, la guía británica 2010 incluye un apartado específico para el cribado y vigilancia de los pacientes con acromegalia, dentro de las poblaciones de riesgo53. Existe poca evidencia científica en este aspecto por lo que es mejor dar recomendaciones basadas en guías clínicas ya que recogen mejor la evidencia científica que los estudios concretos.
Según esta guía, en los pacientes acromegálicos ha de realizarse una colonoscopia basal, completa y de calidad a los 40 años de edad, con controles endoscópicos cada 5 años si las endoscopias son normales54.
Obtener una colonoscopia de calidad es más difícil que en la población general por el hecho de ser frecuente el dolicocolon y tener un tiempo de tránsito superior por lo que algunos autores recomiendan doblar la dosis de solución evacuante55.
Cuando se ha detectado un pólipo adenomatoso en la primera colonoscopia, se recomienda efectuar un seguimiento riguroso mediante exámenes endoscópicos cada dos o tres años, especialmente si este es mayor de 10mm y principalmente en los pacientes que presentan respuesta insuficiente al tratamiento médico56, aunque según la guía británica 2010, las recomendaciones de seguimiento de pólipos son las habituales.
Los niveles elevados de GH y la presencia de pólipos en la colonoscopia basal son factores relacionados con la recurrencia polipoidea56.
Del resto de tumores, no se ha descrito mayor incidencia respecto a la de la población general, no obstante existe un amplio espectro de tumores malignos descritos en la literatura en pacientes acromegálicos17,57-59.
Es infrecuente, pero existen casos de pacientes con acromegalia y múltiples tumores60. Tal es así, que fue a raíz de un caso clínico que manejamos, con acromegalia, swchammoma, seminoma clásico, pólipos adenomatosos colónicos y adenocarcinoma gástrico, que los autores de esta revisión decidimos realizar esta actualización del tema.
La tasa de incidencia estandarizada de neoplasias gastrointestinales a otros niveles también se encuentra aumentada, pero en menor cuantía. La serie más amplia al respecto, sobre una cohorte de 1.041 pacientes acromegálicos9, determina una tasa de cáncer esofágico de 3,1 respecto a la población general, de 2,5 en el cáncer gástrico, y de 6,2 en el intestino delgado.
En el caso del cáncer gástrico, está descrita la sobreexpresión de IGF-I en el tejido tumoral de la pieza quirúrgica61. Este hecho, asociado o no la infección por Helicobacter pylori, podría determinar la aparición de este tipo de tumores a edades más tempranas y con histología más agresiva, como el caso clínico que los autores de esta revisión manejamos.
Diversos estudios sugieren una mayor incidencia de tumoraciones pulmonares, mamarias, tiroideas y prostáticas10–17 aunque esta última es más controvertida y existen estudios que refieren un riesgo similar al de la población general15.
PronósticoEsta enfermedad acorta la esperanza de vida aumentando la mortalidad al doble o al triple de la esperada6.
Las muertes se producen en primer lugar por causa cardiovascular en pacientes menores de 60 años.
Las causas respiratorias suponen el 25% de la mortalidad ocupando los tumores la tercera causa de mortalidad.
La enfermedad asocia también un notable incremento de la morbilidad; la HTA, DM, artrosis y miocardiopatía, específica o no, se presentan con mayor frecuencia que en la población general30.
TratamientoLos principales pilares del tratamiento serían la prevención de la patología neoplásica, especialmente de origen colorrectal y los trastornos cardiovasculares y respiratorios25.
Los objetivos terapéuticos son erradicar el tumor y normalizar la secreción de GH e IGF-I. Todo ello, se debe intentar conseguir preservando la función tiroidea.
La cirugía transesfenoidal está indicada en todos los adenomas hipofisarios, excepto en los que presentan extensión supraselar amplia. En manos expertas, se logra alcanzar la curación con criterios estrictos en el 45-55% de los casos62,63.
Los factores pronósticos que influyen en una resección curativa son el tamaño del tumor, su invasividad y las concentraciones prequirúrgicas de GH64.
La radioterapia estereotáxica fraccionada está indicada cuando tras la cirugía trasesfenoidal no se logra normalizar la GH y muy raramente como terapia primaria65.
El porcentaje de descenso de GH respecto a su concentración basal oscila entre el 42 y 60% a los dos años y el 75-80% a los 5 años. Este lento descenso logra controlar la enfermedad en el 70% de los casos a los 10 años65,66.
Tratamiento médicoLos análogos de la somatostina son actualmente los fármacos más empleados.
Consiguen cifras de GH menores a 2,5μg/l en el 40-50% de los casos y normalizan la IGF-I en el 40% de los casos67.
También inducen una moderada reducción del tamaño del tumor. Son los únicos fármacos efectivos en la acromegalia causada por tumores con secreción ectópica de GHRH estando indicados cuando hayan fracasado otras terapias y para el tratamiento preoperatorio68,69.
Los agonistas dopaminérgicos tienen una efectividad baja, logrando descender la GH<5μg/l solo en el 20% de los casos y normalizar la IGF-I en el 10%; mejoran en todo caso la sintomatología hasta en el 60% de los casos70.
Los bloqueantes de receptores de GH no tienen un uso sistematizado.
ConclusiónEl objetivo terapéutico para disminuir la morbimortalidad de la acromegalia es la curación, o en su defecto, el control de la enfermedad.
La patología tumoral tiene una incidencia variable, pero aumentada respecto a la de la población general. En el caso del CCR, las colonoscopias de cribado están indicadas en el paciente acromegálico a partir de los 40 años de edad, con más de 10 años de evolución de la enfermedad o por antecedentes familiares de CCR.
Dado que el cáncer gástrico presenta también un discreto aumento del riesgo relativo respecto al de la población general, los autores de esta revisión proponemos la realización de gastroscopia y colonoscopia de forma conjunta, al menos en el estudio basal. El coste-beneficio de esta exploración precisa de estudios más amplios.
Finalmente, el mayor conocimiento de la morbimortalidad asociada a esta entidad es la mejor estrategia terapéutica y de seguimiento, adelantándonos en muchos de los casos al curso evolutivo esperable.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

