




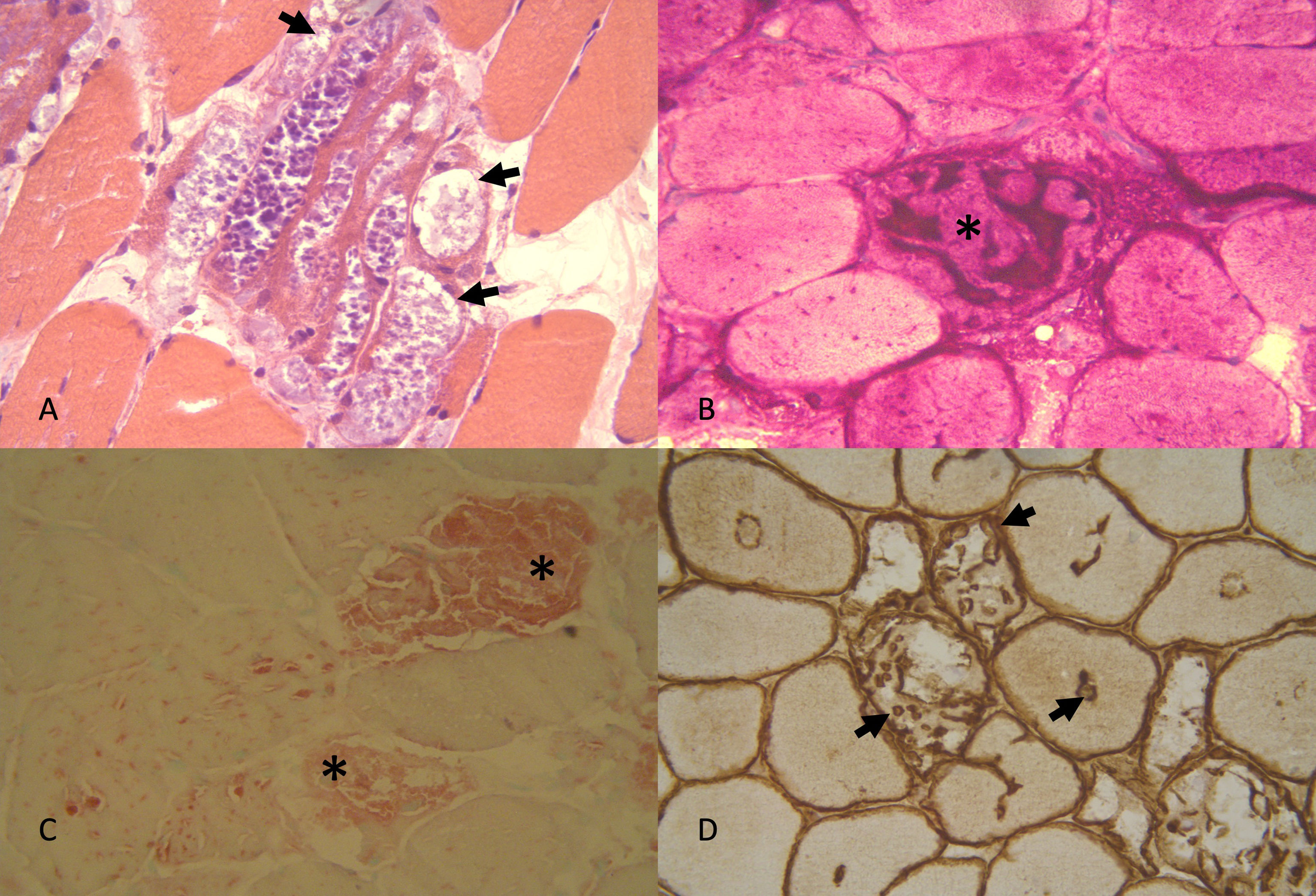
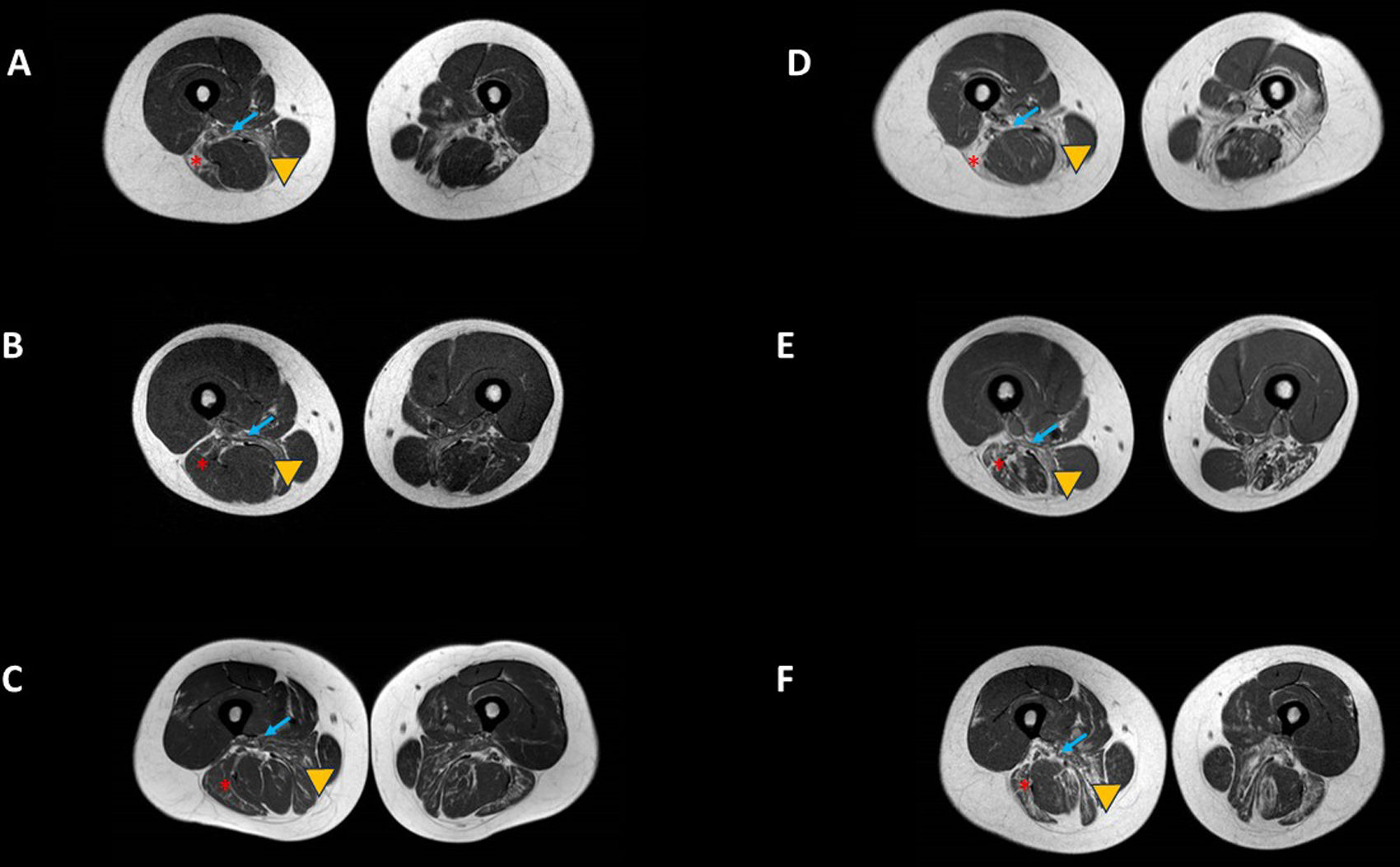
La enfermedad de Pompe (EP) es una enfermedad lisosómica debida a un déficit de la enzima alfa-glucosidasa ácida (GAA), que se manifiesta fundamentalmente como una miopatía progresiva con afectación respiratoria temprana. Desde el año 2006 existe un tratamiento de reemplazo enzimático (TRE).
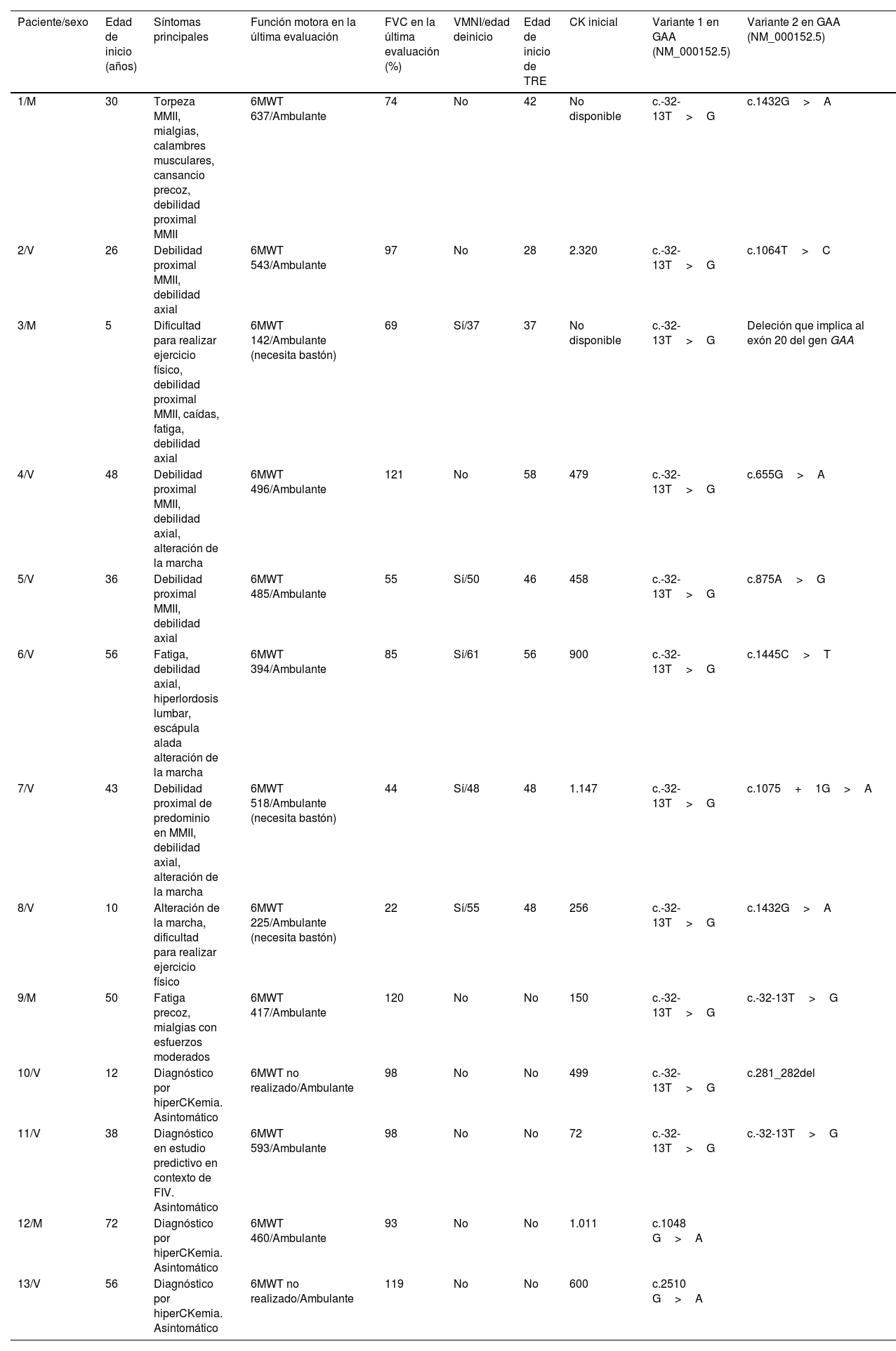
Material y métodosSe describen los casos de 13 pacientes con déficit parcial de la GAA en seguimiento en el Hospital 12 de Octubre, 8 de ellos en tratamiento.
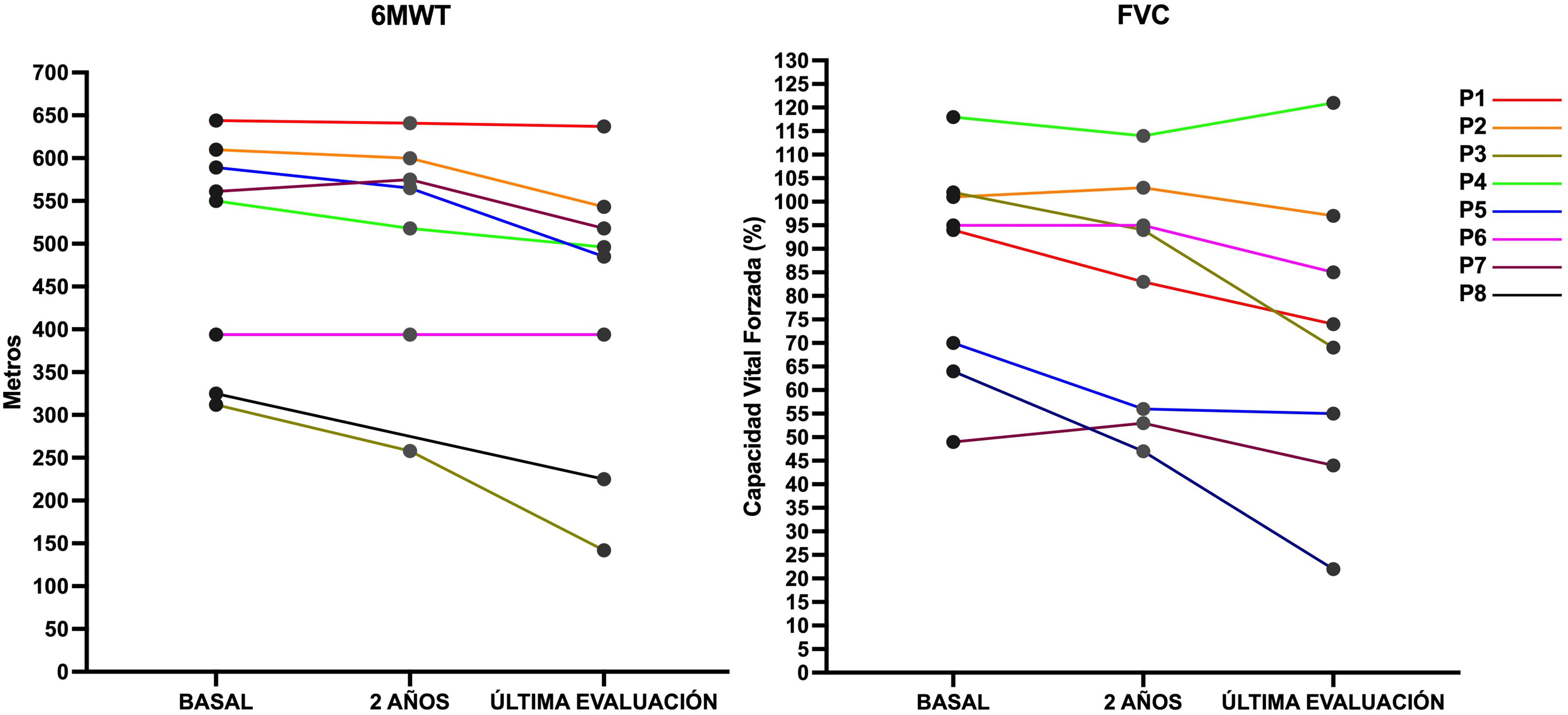
ResultadosOcho de los pacientes tienen síntomas, todos ellos de inicio tardío. Presentan debilidad axial y proximal de predominio en extremidades inferiores, pero conservan la marcha autónoma. Cinco pacientes requieren ventilación mecánica no invasiva por insuficiencia respiratoria. Todos los pacientes con síntomas reciben TRE y en 7/8 (87,5%) existe un empeoramiento de la función motora y pulmonar tras una media de 8,25 años de tratamiento (FVC y 6MWT, media basal y postratamiento 86,6% vs. 79,8% y 498 vs. 430m, respectivamente).
ConclusiónNo todos los pacientes con déficit parcial de GAA experimentan síntomas de EP y los sintomáticos, a pesar la TRE, experimentan en su mayoría un deterioro motor y respiratorio gradual.
.
Pompe Disease (PD) is a lysosomal disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA), primarily manifesting as a progressive myopathy with early respiratory involvement. Enzyme replacement therapy (ERT) is available since 2006.
Materials and methodsWe describe 13 patients with partial GAA deficiency, followed at Hospital 12 de Octubre, 8 of whom were receiving treatment.
Results8 patients exhibit symptoms, all with late onset. They display axial and proximal weakness predominantly in the lower limbs but maintain autonomous gait. Five patients require non-invasive mechanical ventilation due to respiratory insufficiency. All symptomatic patients receive ERT, and in 7/8 (87.5%), there is a decline in motor and pulmonary function after an average of 8.25 years of treatment (baseline and post-treatment FVC and 6MWT mean 86.6% vs 70.8% and 498 vs 430 meters, respectively).
ConclusionNot all patients with partial GAA deficiency experience symptoms of PD, and symptomatic patients, despite ERT with recombinant alpha-glucosidase, mostly experience a gradual decline in motor and respiratory function.

