




Fabry disease (FD) is an X-linked lysosomal storage disorder due to a deficiency of the α-galactosidase A enzyme. Although women were historically considered only carriers, many studies have contradicted this fact. The main aim of this work was to set the first Spanish study out of the on-going registries on health status and management of women diagnosed with FD who were not receiving enzyme replacement therapy (ERT).
Material and methodsAn epidemiological, cross-sectional, descriptive and multicentre study was assessed in women diagnosed for FD who were not receiving ERT. Assessments on symptomatology and severity were collected using several clinical questionnaires. Additionally, clinical information and lab tests were obtained from clinical records.
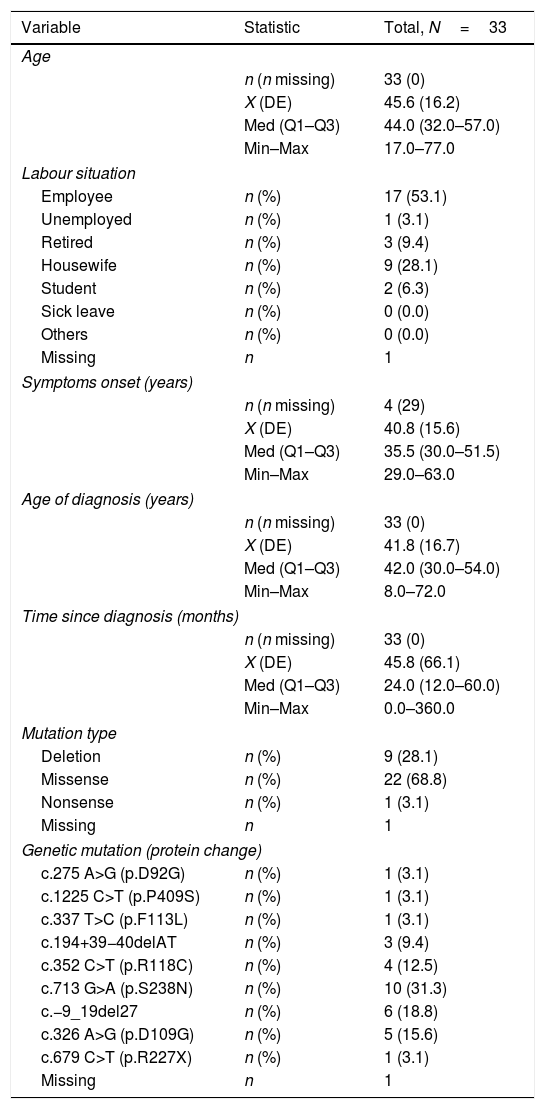
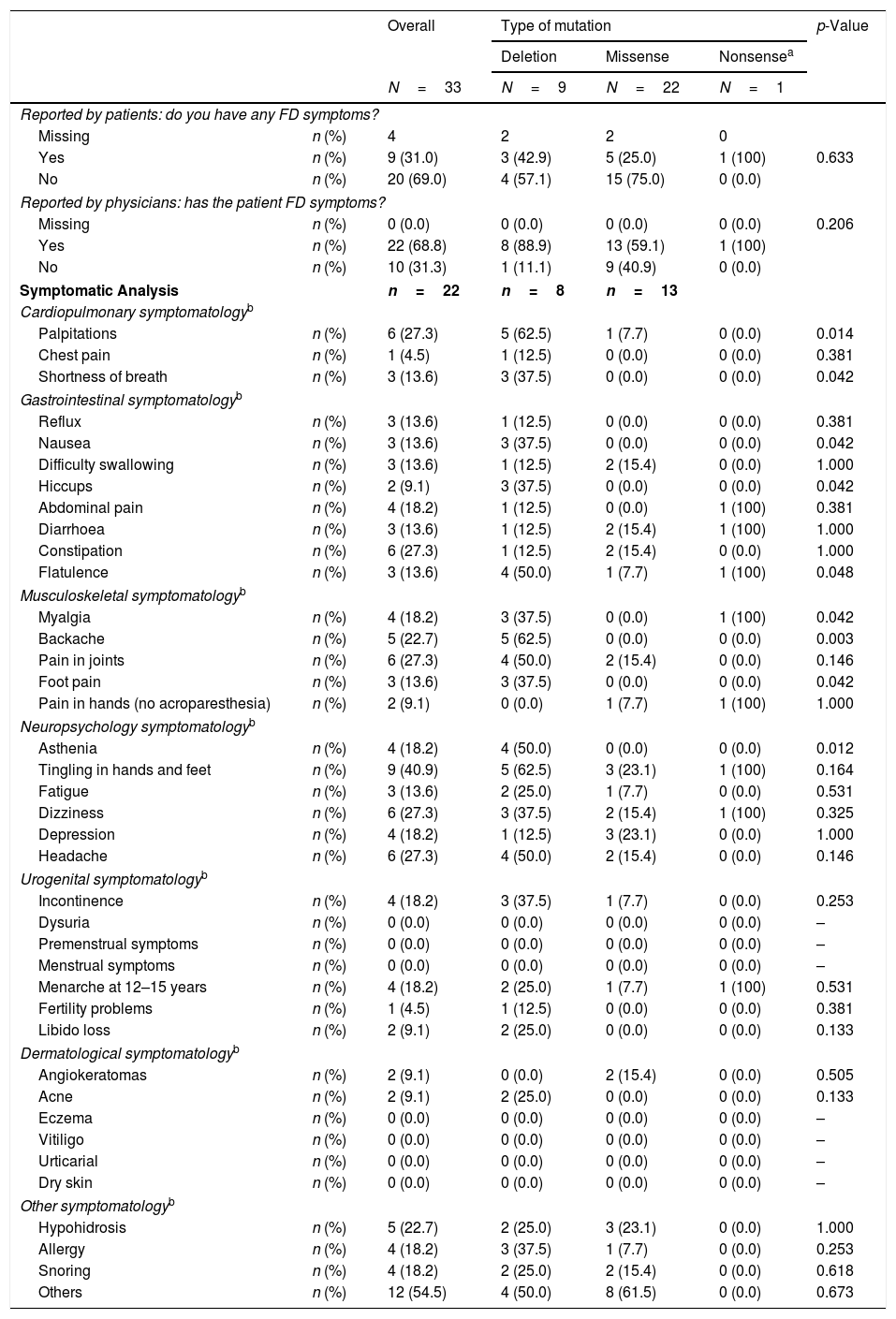
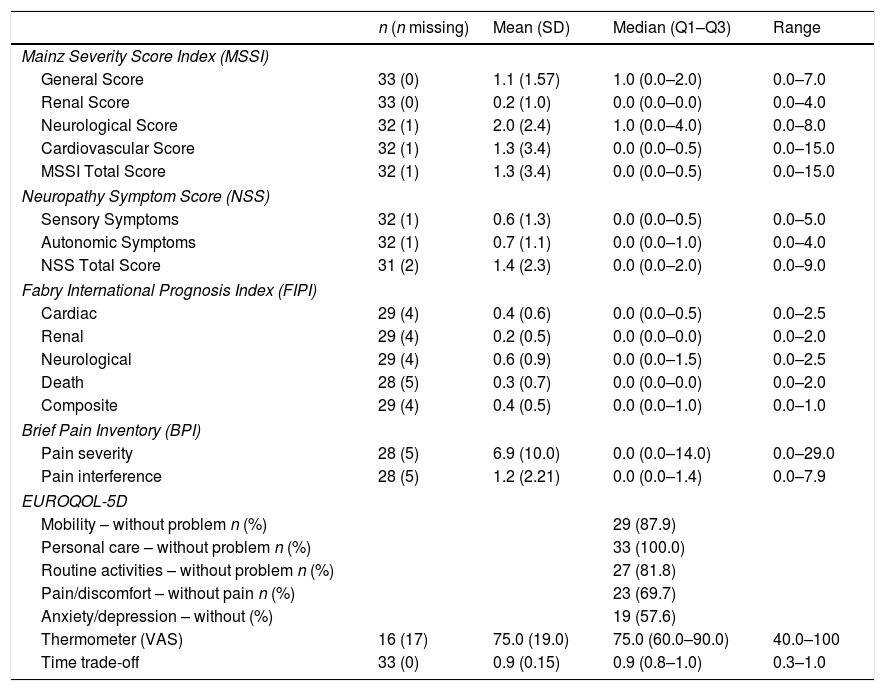
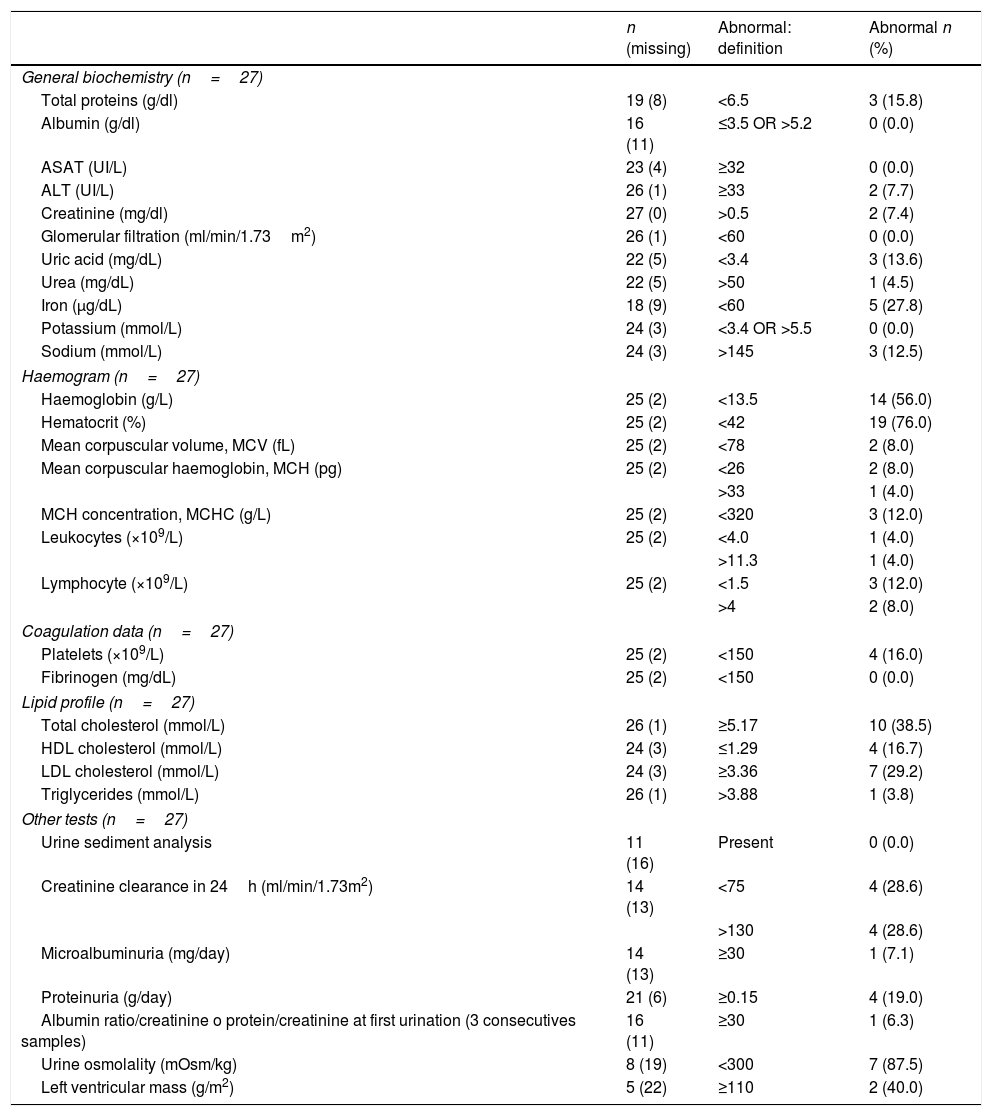
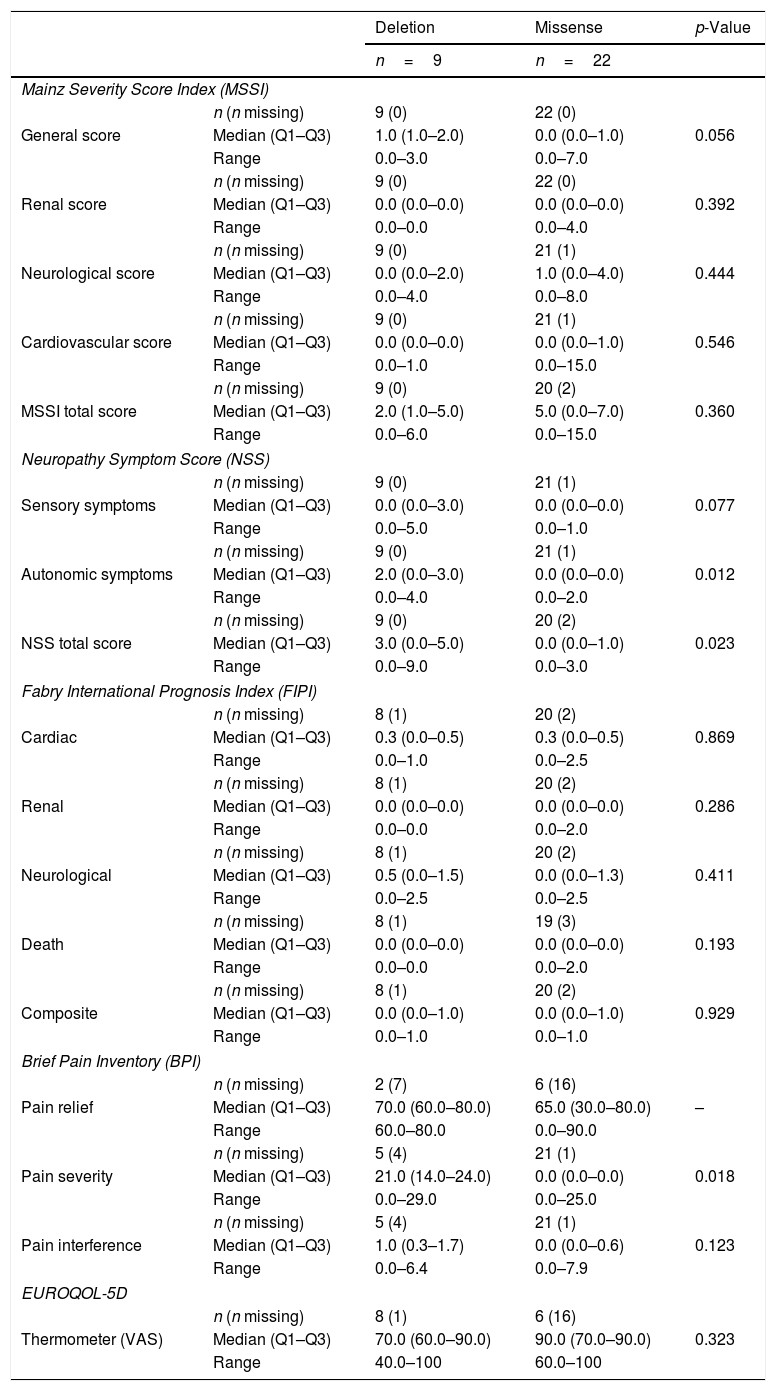
ResultsThirty-three women with a mean age of 45.6±16.2 years were studied. Symptom onset was at a median age of 35.5 years old (range: 30.0–51.5), being diagnosed a median of 2 years later (range: 1.0–1.5). Missense mutations were the most prevalent mutation (n=22, 68.8%). Although 69% considered themselves as asymptomatic, 22 (66.7%) showed at least one FD-related clinical symptom. Using Mainz Severity Score index and Fabry International Prognosis Index neurological symptomatology obtained higher scores both for severity and prognostic. The EQ-5D questionnaire showed 42.2% patients referring to some anxiety or depression, and 30.3% thought that their life was somehow altered by the pain. 62.5% were not receiving any treatment and ERT was offered only to one patient (3.6%) who refused it.
ConclusionsAlthough most of the heterozygous women for FD had not received ERT or either symptomatic treatment, they present symptoms of disease. Careful follow-up of female patients or some adjuvant treatment may be considered to delay progressive organ damage and improve patient quality of life.
La enfermedad de Fabry (EF) es un trastorno de almacenamiento lisosómico hereditario, ligado al cromosoma X y derivado de una deficiencia de la enzima α-galactosidasa A. Aunque históricamente solo se ha considerado portadoras a las mujeres, esto ha sido contradicho por muchos estudios. El objetivo principal de este trabajo ha sido establecer un primer estudio español independiente de los registros actuales sobre la situación y seguimiento clínico de las mujeres diagnosticadas con EF que no recibían terapia de sustitución enzimática (TRE).
Material y métodosSe llevó a cabo un estudio epidemiológico, transversal, descriptivo y multicéntrico en mujeres diagnosticadas con EF que no recibían TRE. Las evaluaciones sobre la sintomatología y la gravedad fueron recopiladas mediante varios cuestionarios clínicos. Adicionalmente se obtuvo información clínica y resultados de pruebas de laboratorio de las historias clínicas.
ResultadosSe estudiaron 33 mujeres con una edad media de 45,6±16,2 años. El inicio de los síntomas se produjo a una mediana de edad de 35,5 años (rango: 30,0-51,5), siendo diagnosticado en una mediana de 2 años después (rango: 1,0-1,5). Las mutaciones de sentido erróneo fueron las más frecuentes (n=22; 68,8%). Aunque el 69% se consideraron a sí mismas asintomáticas, 22 (66,7%) mostraron al menos un síntoma clínico relacionado con la EF. Utilizando el índice de severidad de Mainz y el índice pronóstico internacional de Fabry, la sintomatología neurológica obtuvo puntuaciones más altas tanto para la gravedad como para el pronóstico. El cuestionario de calidad de vida EQ-5D mostró que el 42,2% de las pacientes referían cierta ansiedad o depresión, y el 30,3% pensó que su vida estaba interferida de alguna manera por el dolor. El 62,5% no recibía ningún tratamiento y solo se ofreció TRE a una paciente (3,6%), que lo rechazó.
ConclusionesAunque la mayoría de las mujeres heterocigotas para la EF no habían recibido TRE, ni tampoco ningún tratamiento sintomático, sí presentan síntomas de la enfermedad. Un seguimiento cuidadoso de las pacientes junto con alguna terapia adyuvante podría ser de interés para retrasar el daño progresivo de los órganos y mejorar la calidad de vida de las pacientes.

