




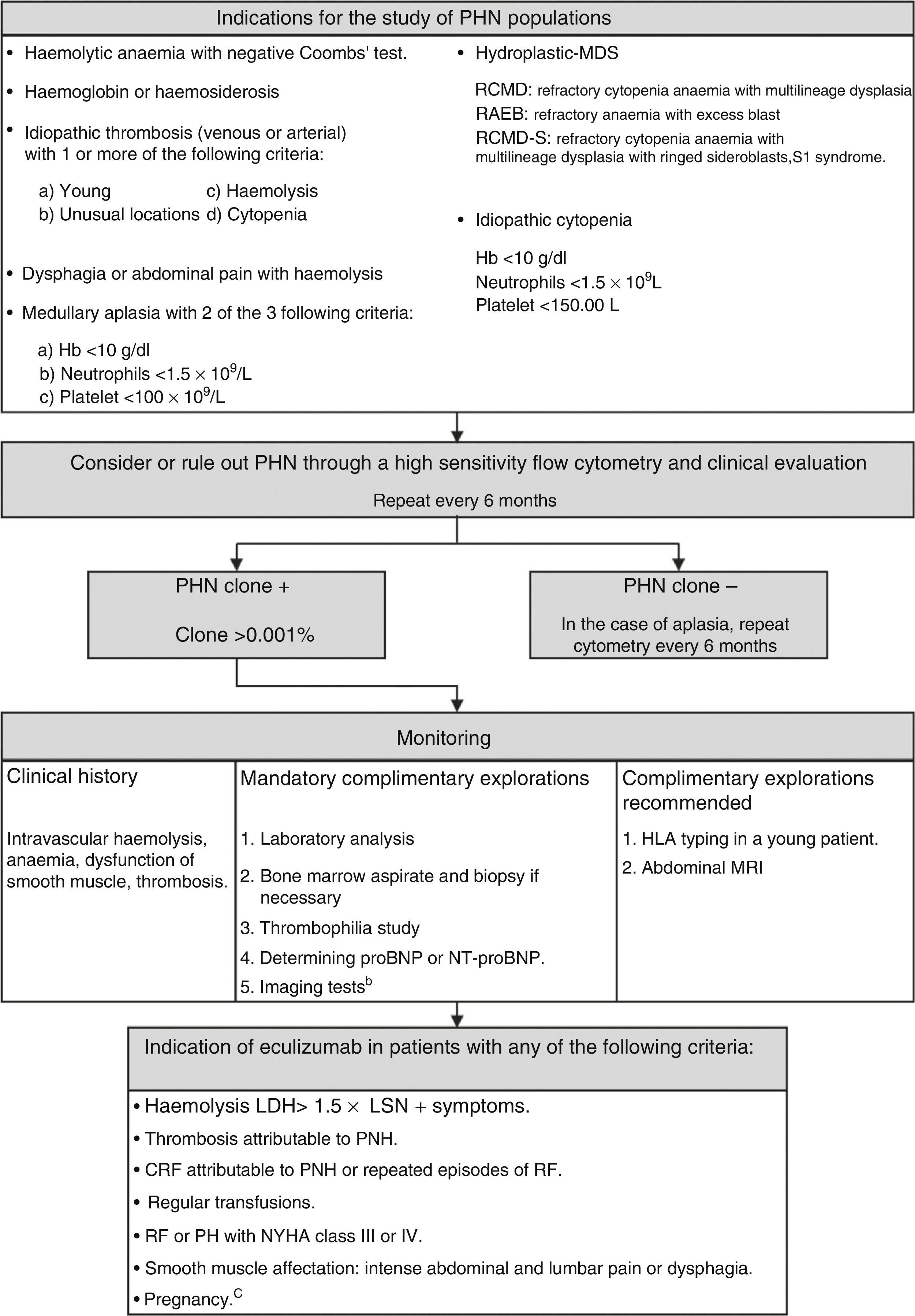

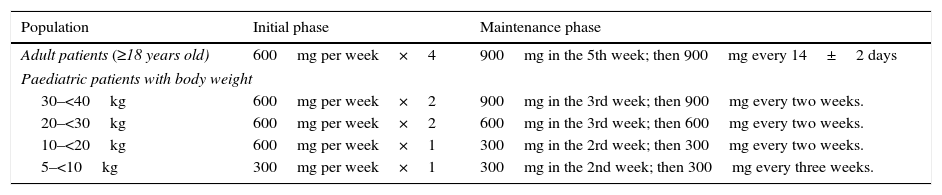
Paroxysmal nocturnal haemoglobinuria (PNH) is an acquired clonal disorder of the haematopoietic progenitor cells due to a somatic mutation in the X-linked phosphatidylinositol glycan class A gene. The disease is characterised by intravascular haemolytic anaemia, propensity to thromboembolic events and bone marrow failure. Other direct complications of haemolysis include dysphagia, erectile dysfunction, abdominal pain, asthenia and chronic renal failure (65% of patients). The disease appears more often in the third decade of life and there is no sex or age preference. Detection of markers associated with glucosyl phosphatidyl inositol deficit by flow cytometry is currently used in the diagnosis of PNH. For years, transfusions have been the mainstay of therapy for PNH. A breakthrough in treatment has been the approval of the humanised monoclonal antibody eculizumab, which works by blocking the C5 complement protein, preventing its activation and therefore haemolysis. Several studies have confirmed that treatment with eculizumab avoids or decreases the need for transfusions, decreases the probability of thrombosis, improves the associated symptomatology and the quality of life in patients with PNH, showing an increase in survival. Because of rapid advances in the knowledge of the disease and its treatment, it may become necessary to adapt and standardise clinical guidelines for the management of patients with PNH.
La hemoglobinuria paroxística nocturna (HPN) es una enfermedad clonal de las células progenitoras hematopoyéticas originada por la mutación adquirida del gen fosfatidil-inositol-glicano del grupo A, situado en el brazo corto del cromosoma X. Se caracteriza por anemia hemolítica intravascular, tendencia a la trombosis y un componente variable de insuficiencia medular. Otras complicaciones derivadas de la hemólisis son disfagia, disfunción eréctil, dolores abdominales, astenia e insuficiencia renal crónica (un 65% de los pacientes). La enfermedad afecta por igual a ambos sexos y puede aparecer a cualquier edad, con una mayor incidencia en la tercera década de la vida. Actualmente, el diagnóstico se basa en la detección de poblaciones celulares con marcadores asociados al déficit de glucosil-fosfatidil-inositol mediante citometría de flujo. Durante años, el pilar terapéutico de la HPN hemolítica era el soporte transfusional. Un gran avance en el tratamiento ha sido la aprobación del anticuerpo monoclonal humanizado eculizumab, que bloquea la proteína C5 del complemento impidiendo su activación, y por tanto, la hemólisis. Diversos estudios han confirmado que el tratamiento con eculizumab evita o disminuye el requerimiento transfusional, reduce la probabilidad de trombosis, mejora la sintomatología asociada y la calidad de vida de los pacientes con HPN, mostrando un aumento de la supervivencia. Este rápido avance en el conocimiento de la enfermedad y su tratamiento hace necesario adaptar y homogeneizar las directrices de actuación clínica en el manejo de pacientes con HPN.

