


dysostosis spondylothoracic, or Jarcho Levin syndrome, is characterized by a short neck and thorax, a protruding abdomen, abnormal vertebral segmentation and fusion posterior costal resulting in thoracic restriction or respiratory failure and scoliosis. The prevalence is estimated at 1 in 12,000 live births for the people of Puerto Rico and 1 per 200,000 for the rest of the world. It is inherited in an autosomal recessive manner and the only related gene is MESP2.
Clinical caseNewborn male, who during the first hour of life develops perioral cyanosis, thoracoabdominal dissociation and polipnea, requiring endotracheal intubation and mechanical ventilation for respiratory impairment, finding thoracoabdominal costovertebral abnormalities with an x-ray, and a conditioning restrictive pattern like a crab. During the physical examination, we found horizontal eyelid openings, right atrial appendage, straight nasal bridge, short thorax and asymmetry and hypertrichosis, predominantly in the back. A diagnosis of dysostosis spondylothoracic is confirmed, and the patient was discharged at 7 days of age, with follow up neonatal consultation at high risk.
ConclusionIn a neonate with respiratory distress syndrome, costovertebral assessment becomes important, with the intention of discarding syndromes associated with defects in the costovertebral segmentation, as Jarcho Levin syndrome, which causes respiratory impairment that can lead to respiratory failure and death.
Spondylothoracic dysostosis, also known as Jarcho Levin syndrome, is characterized by a short neck and thorax, a protruding abdomen, inguinal and umbilical hernias, abnormal vertebral segmentation and costal fusion resulting in thoracic restriction or respiratory failure, urinary tract anomalies and scoliosis, which is not common, yet it may be severe if it occurs.1,2
Prevalence in the population of Puerto Rico is estimated at 1 per 12,000 live births. The number is not exact for the rest of the world. However, calculations suggest it occurs in 1 in 200,000 live births.2
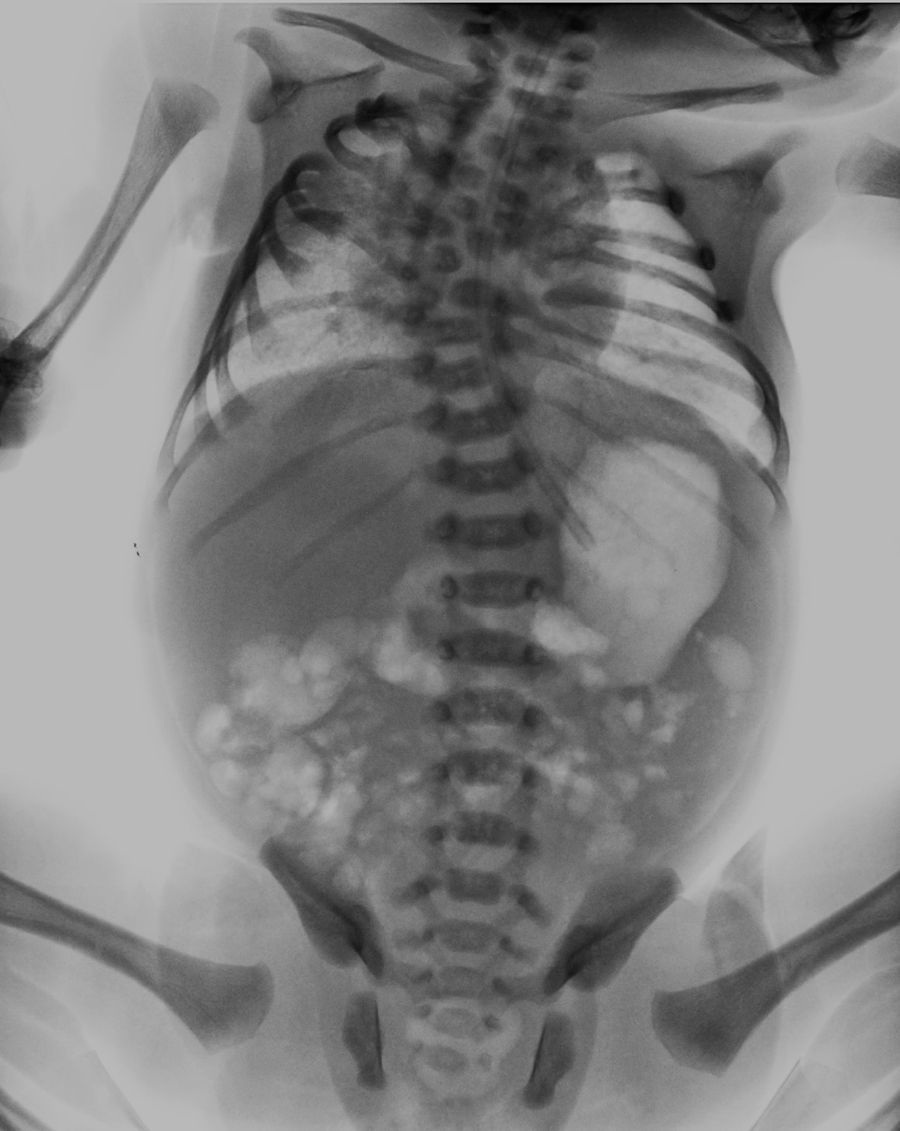
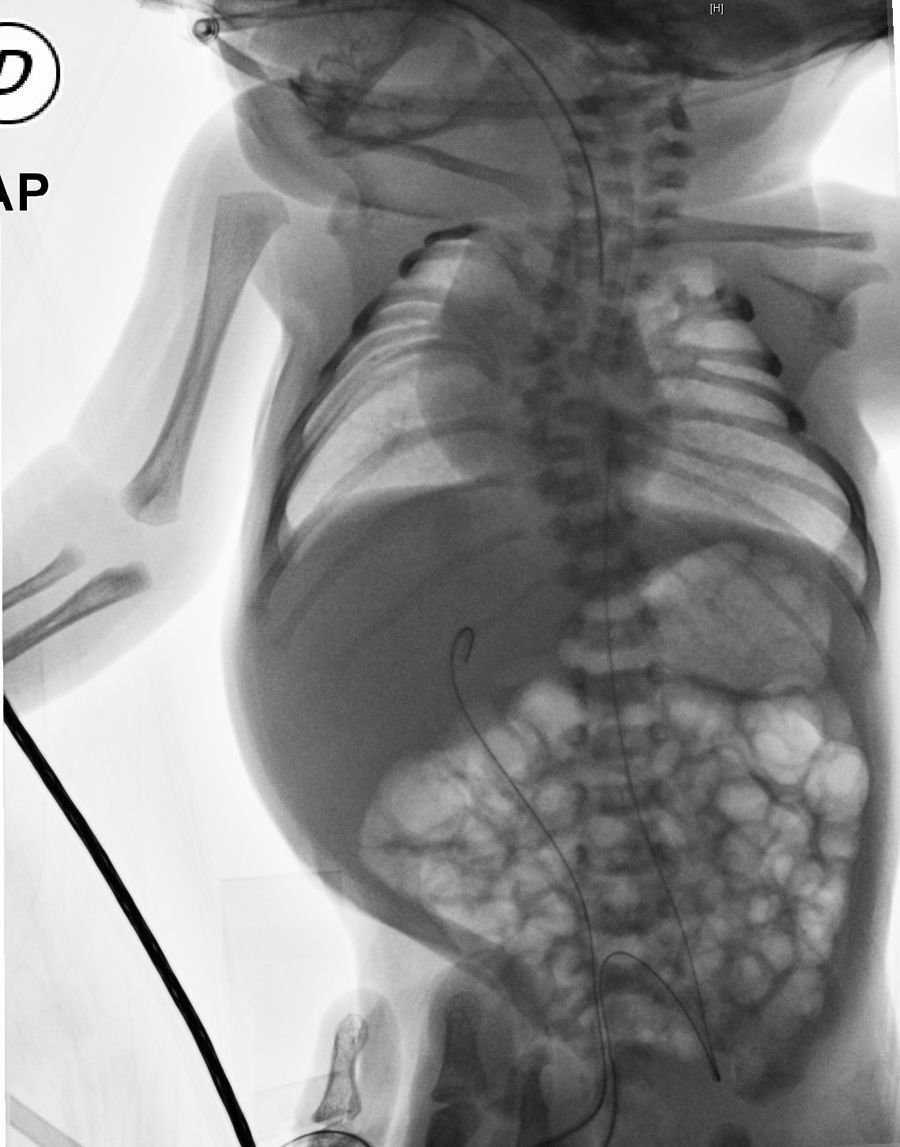
Distinctive radiographic findings include: abnormal segmentation of all vertebral segments with “H-shaped vertebrae” and severe shortening of the spine, especially in the dorsal area. The anteroposterior radiograph displays fan-shaped ribs from the costovertebral base, giving it a “crab-like” aspect. A distinctive characteristic is the “tramline sign”, which results from an early prominence in the vertebral pedicles, in contrast to the vertebrae bodies, which do not possess a regular shape.1–3
Costal and vertebral alterations lead to a significant thoracic restriction in 60% of newborns, resulting in respiratory distress syndrome and requiring immediate medical intervention. Because of the extensive costal fusion, the intercostal muscles are not able to expand the thorax. A pulmonary hypoplasia syndrome is described due to the reduced size of the thorax; however, no intrinsic pulmonary alteration has been described. As a result of the respiratory complication, newborns have a mortality rate of up to 40%. Approximately 90% of patients develop inguinal hernias, bilaterally in 75% of cases. Umbilical hernias occur in 15% of cases; these occur due to the pressure increase in the abdominal cavity as a result of an excessive use of the diaphragm during breathing.1,3,4
Physical characteristics include a prominent occiput in newborns with a posterior flattening, giving it the appearance of brachycephaly. Posterior hair implantation line is low, with a prominent nose bridge in 33% of cases, a normal philtrum in length and shape, and a high palate in 75% of cases. Cardiac anomalies are rare. Atrial septal defects are the least common malformation, occurring in less than 5% of cases. Other congenital anomalies include clubfoot (1%), cleft palate (>1%), double collecting system (1%) and unilateral glenoid agenesis (0.5%).1–4
The secondary most important complication is chronic respiratory failure, which is caused by the reduced lung capacity. This may result in pulmonary hypertension and cardiac failure.5
Case presentationNewborn male, full-term 37.5 weeks of gestation using the Capurro method; his mother, 18 years old and his father 22, apparently healthy, non-blood relatives, without exposure to teratogens, product of a first gestation, with adequate prenatal care, 8 obstetric ultrasounds with no alterations, with 2 episodes of urinary tract infection during the first trimester treated with antibiotic therapy without complications, with iron, folic acid and multivitamin intake starting on the second month of pregnancy. The baby is born in a private clinic, via elective C-section at 37.5 weeks of gestational age, weighing 2780g, with a cephalic perimeter of 35cm and an Apgar score of 8/8. During the first hours of life the baby presents peribuccal cyanosis, thoracoabdominal dissociation and polypnea of 80–90 breaths per minute, a cephalic face mask is placed with fio2 at 100%. The cyanosis and throracoabdominal dissociation disappeared, reducing fio2 to 60% with the persistency of polypnea; therefore, he is referred to our hospital in his first day of life for diagnosis and management.
His physical examination showed horizontal palpebral fissures, right atrial appendage, straight nose bridge, asymmetrical thorax, telethelia, no murmurs, no masses or visceromegaly were found, hypertrichosis mainly on the back, and the presence of intercostal and subcostal retractions and polypnea.
Umbilical venous and arterial catheters are placed and a thoracoabdominal radiograph is taken, observing costovertebral alterations which condition the presence of a restrictive pattern, cervical and dorsal hemivertebra, accentuated scoliosis, absence of the 12th right rib and the 11th and 12th left ribs with bilateral asymmetric costal fusion (Figs. 1 and 2). On the third day of life, he presented respiratory clinical deterioration with respiratory acidosis, requiring endotracheal intubation and mechanical ventilation for 24h, accomplishing extubation to a cephalic face mask with an improvement in the respiratory pattern. The oral route is initiated on his fourth day of life, with the mother's breast through an orogastric tube, allowing progress and offering feeding through section with an adequate tolerance. An echocardiogram is performed as part of the approach, finding a small oval foramen and a transfontanellar ultrasound without alterations. Due to findings, during the physical examination and the presence of costovertebral alterations in the thoracoabdominal radiograph, a referral to the Medical Genetics Service is made. They conduct a genetic clinical history and a detailed clinical evaluation of the patient, giving him a diagnosis of spondylothoracic dysostosis. He is admitted to the Neonatal Intermediate Care Unit for maternal training and pediatric orthopedic assessment for the planning of costovertebral alteration management. He is released from the hospital on day 7 of his extrauterine life, with a reassessment of the high-risk and medical genetics neonatal follow-up consultation.
Discussion

Spondylothoracic dysostosis in its neonatal stage produces respiratory clinical secondary to the presence of a short thorax in up to 65% of newborns, with abnormal vertebral segmentation and posterior costal fusion facilitating the presence of a restrictive breathing pattern capable of causing anything from mild distress to a full uncompensated respiratory acidosis requiring endotracheal intubation and mechanical ventilation, which once initiated, its early removal should be procured. The use of pulmonary surfactant may sometimes be necessary based on neonatal respiratory distress protocols, continuous monitoring of the cardiorespiratory constants and intensive management of infectious diseases. During the early stages of childhood, early detection and management of respiratory infections are of great importance, as well as immunization against respiratory syncytial virus and considering management with bronchodilator-based therapy in patients with intercurrent conditions.1,2,5
Our patient's respiratory distress syndrome evolved as described by the literature going from mild distress to a respiratory distress requiring endotracheal intubation. However, it is worth noting the importance of costovertebral radiological findings and their integration, as well as the physical examination of Jarcho Levin syndrome.
Clinical knowledge of spondylothoracic dysostosis, or Jarcho Levin syndrome, is of great importance, since it usually manifests with a respiratory distress syndrome at birth, frequently leading to pre-term and term newborns being admitted to the hospital. A detailed assessment of the thoracoabdominal radiograph used as an adjunctive initial diagnostic tool in all newborns with respiratory distress provides radiological data which are characteristic of the disease, contributing to its early identification and favoring its optimal treatment and approach.
Recognizing these characteristics clearly differentiates it from the rest of the pulmonary diseases coursing with respiratory distress syndrome in newborns. Moreover, knowing the behavior of the disease and its management improves survival and the patient's quality of life during follow-up at the pediatric age.
Pediatric surgery and orthopedic assessments are necessary during patient follow-up subsequent to acute event resolution of the surgical planning of scoliosis, costal fusions, inguinal and umbilical hernia,6 though malformation treatment is conservative in most cases, with periodical radiographic controls, physiotherapy and infection control.
Regarding the genetic aspect, it is important to take into account several factors, such as a family history, focusing on skeletal dysplasia, consanguinity, Spanish or Puerto Rican lineage, and evaluation of radiological findings in search of costovertebral alterations characteristic of spondylothoracic dysostosis.
It is inherited in a recessive autosomal form. The only gene associated with spondylothoracic dysostosis is MESP2. When sequencing the gene, we are able to find 3 mutations: Gly103*, p.Leu125Val and p.Glu230*.1
In this case, since both parents are phenotypically healthy and there is no other family history, we are able to infer the risk of recurrence for the parents based on the fact that they carry a heterozygous mutation (that is, they present only one mutant allele). Thus, for every pregnancy, they would have a 25% chance of having an affected baby, a 50% chance for them to be asymptomatic carriers just like their parents, and a 25% chance of being healthy. The patient's children will inevitably be asymptomatic carriers (in case they had children with a healthy person). It is worth noting that a molecular study was not conducted in this particular case due to the parents’ economic situation.
Prenatal diagnosis in high-risk pregnancies may be given to the couple in the form of a mutation analysis when it is already known, by amniocentesis or chorionic villus sampling (weeks 10–12). Another available option is preimplantation genetic diagnosis.2,7
It is generally possible to find segmentation and vertebral formation defects in the ultrasound of the second trimester of pregnancy, which may cause suspicion based on the findings of spondylothoracic dysostosis.2,7
It is vital for pediatricians and neonatologists to know the less frequent pathologies in newborns which course with respiratory distress syndrome, such as spondylothoracic dysostosis, since the lack of ability or skill to recognize as well as to integrate radiological data and physical examination may result in a late detection of the illness, thus reducing the survival rate in these types of patients. Assessment by different specialties is of great value, as conducted in this case with the medical genetics service, which may help reaching a timely diagnosis.
Conflict of interestThe authors have no conflicts of interest to declare.