


Sweet syndrome is a rare neutrophilic dermatosis consisting in the onset of high fever, neutrophilia, and typical painful skin lesions including erythematous papules, nodules, and plaques on the face, trunk, and extremities, with a bilateral and asymmetrical pattern. Sweet syndrome is classified as idiopathic, predominating in women; malignancy-associated, mainly with hematological cancer, and drug-induced.
The diagnosis is based on clinical history and skin manifestations, being confirmed by a complete blood count showing neutrophilic leukocytosis, and specific findings in the skin biopsy.
We report the case of a 68 year-old man with a 10-year evolution of dermatomyositis complicated by lung fibrosis, followed 8 years later by non-Hodgkin lymphoma (NHL) accompanied by worsening of his fibrosis. Two years after the successful treatment of NHL the patient developed an acute episode of severe dyspnea, multiple skin lesions, and 95% neutrophilia. At that time the patient had a severe lung function impairment complicated by nosocomial pneumonia that led to his death, a few days after the diagnosis of Sweet syndrome was established by histo-pathology examination.
Sweet syndrome is a rare dermatologic entity that can appear several years after diseases characterized by immune dysfunction such as dermatomyositis and NHL.
Introduction
Sweet syndrome was first described in 1964 by Robert Douglas Sweet.1 In 1986 Su and Liu postulated the criteria for its diagnosis.2 There is no racial predilection, and it is more frequent in women (4:1).3-5
The disease consists of high fever, neutrophilia, and typical painful skin lesions including erythematous papules, nodules, and plaques on the face, trunk, and extremities, with a bilateral and asymmetrical pattern.6,7
Sweet syndrome is classified as: a) idiopathic, which predominantly affects women between 30 and 50 years of age;6,8 b) malignancy-associated, which appears to affect men and women equally; in about 20% of the cases the syndrome is associated with a malignancy, most often acute leukemia;9,10 c) drug-induced, which may occur as a reaction to a medication, most frequently to granulocyte colony-stimulating factor (G-SCF).11-14
The diagnosis is confirmed by a complete blood count (CBC) showing leukocytosis with a predominance of neutrophils, and a skin biopsy demonstrating edema of the papillary dermis and a dense inflammatory leukocyte infiltrate in the lower dermis.5,6,15
The treatment includes prednisolone, or potassium iodide, colchicine, and Lugol's solution.5,6,16 Usually recovery is complete with no recurrence.
Case report
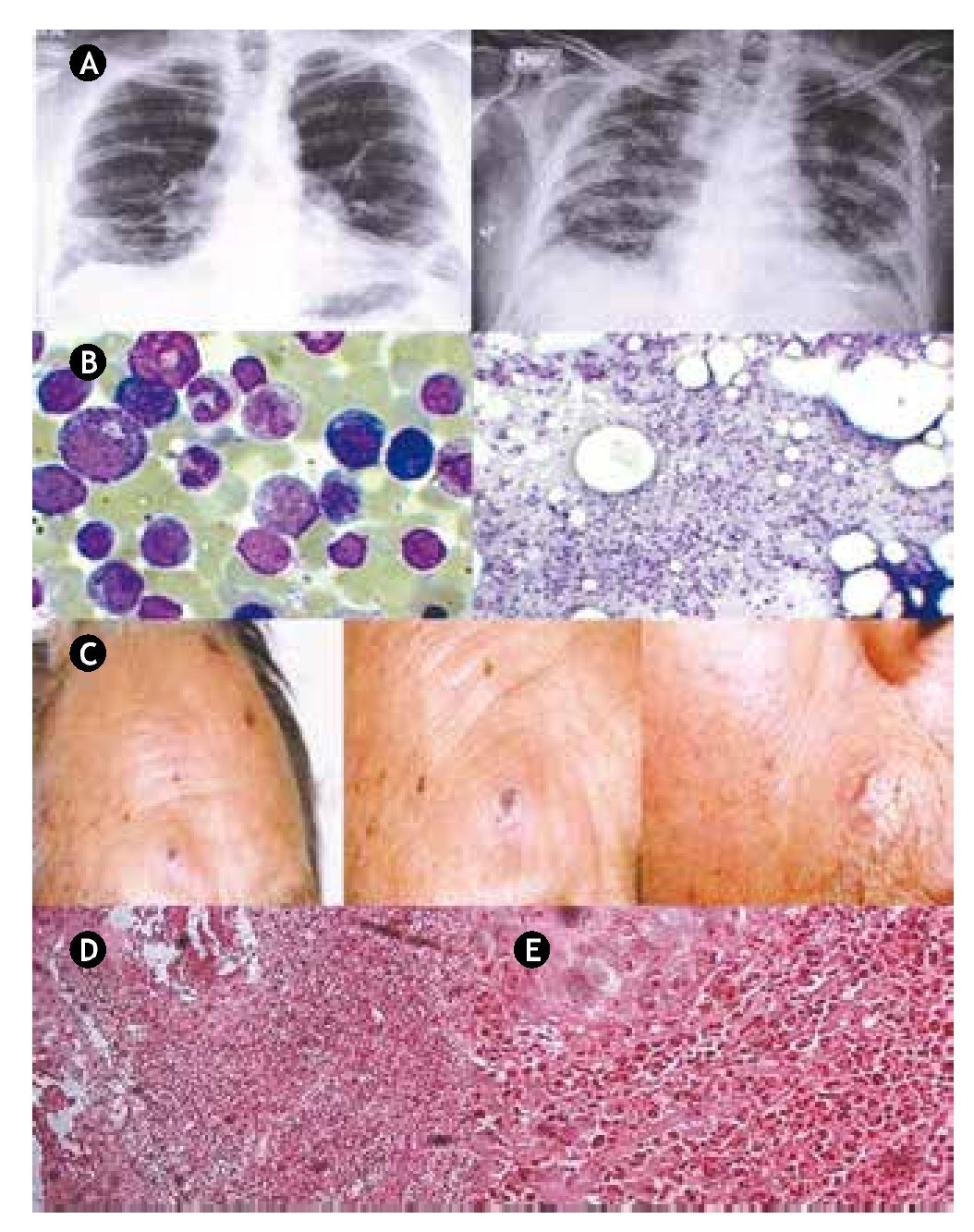
The patient, a 68-year-old man, first showed symptoms in April 2000 with minimum effort dyspnea, accompanied by constitutional symptoms and generalized myalgias with weakness and muscular fatigue. At the time the CBC was normal, but blood chemistry results showed diverse abnormalities including albumin 1.7 g/dl, globulin 4.1 g/dl, lactic dehydrogenase 688 U/l, creatine kinase 7,402 U/l, alanine aminotransferase 283 U/l and aspartate aminotransferase 521 U/l. Chest X-rays showed a bibasal reticular infiltrate (Fig. 1, panel A). Computed tomography (CT) imaging documented interstitial fibrosis in the descamative phase. Spirometry reported a mild to moderate restrictive process. Electromyography of the 4 extremities reported myopathic inflammatory pattern. A muscle biopsy of the quadriceps was normal. Diagnosis of dermatomyositis with secondary lung fibrosis was established and treatment with prednisone and methotrexate was started.
In 2008, physical examination revealed a 12 x 6 cm mass of coalescent adenopathies, firm, nontender, fixed to deep planes in the parotid gland, as well as cervical, retroauricular, bilateral axillary and inguinal lymphadenopathies; in addition, the patient presented B symptoms; there was no visceral enlargement. An excisional biopsy of the axillary and left groin lymph nodes was carried out, leading to the diagnosis of high-grade diffuse large cell non-Hodgkin lymphoma (NHL) with a B immunophenotype. Bone marrow aspirate showed a moderate granulocytic hyperplasia (Fig. 1, panel B). Treatment included rituximab, cyclophosphamide, doxorubicin, and vincristine.
The patient had no major complications until November 2010, when he was admitted to the emergency room after 3 weeks of experiencing malaise, fatigue, and weakness, coupled with the presence of disseminated macular dermatosis, which prompted a consultation to a dermatologist; at that time the patient had painful erythematous papules both over the dorsum of his forearm and face. The lesions rapidly progressed to become nodules and a few of them coalesced forming plaques (Fig. 1, panel C). A skin punch biopsy performed on a left cheek lesion, demonstrated prominent infiltration of leucocytes into the dermis, consistent with the diagnosis of sweet syndrome. (Fig. 1, panels D and E). A CBC reported 10.9 x 109/l WBC, and neutrophilia of 10.3 x 109/l, representing 94.7% of total leukocytes. During his stay at the hospital, the patient developed nosocomial pneumonia that progressed to respiratory failure, and broad spectrum antibiotics were started; a CT scan of the chest showed diffuse bilateral infiltrates, as well as significant progression of pulmonary fibrosis. The ventilatory function worsened, leading to a cardiorespiratory arrest and death of the patient a week after his admission to the hospital.
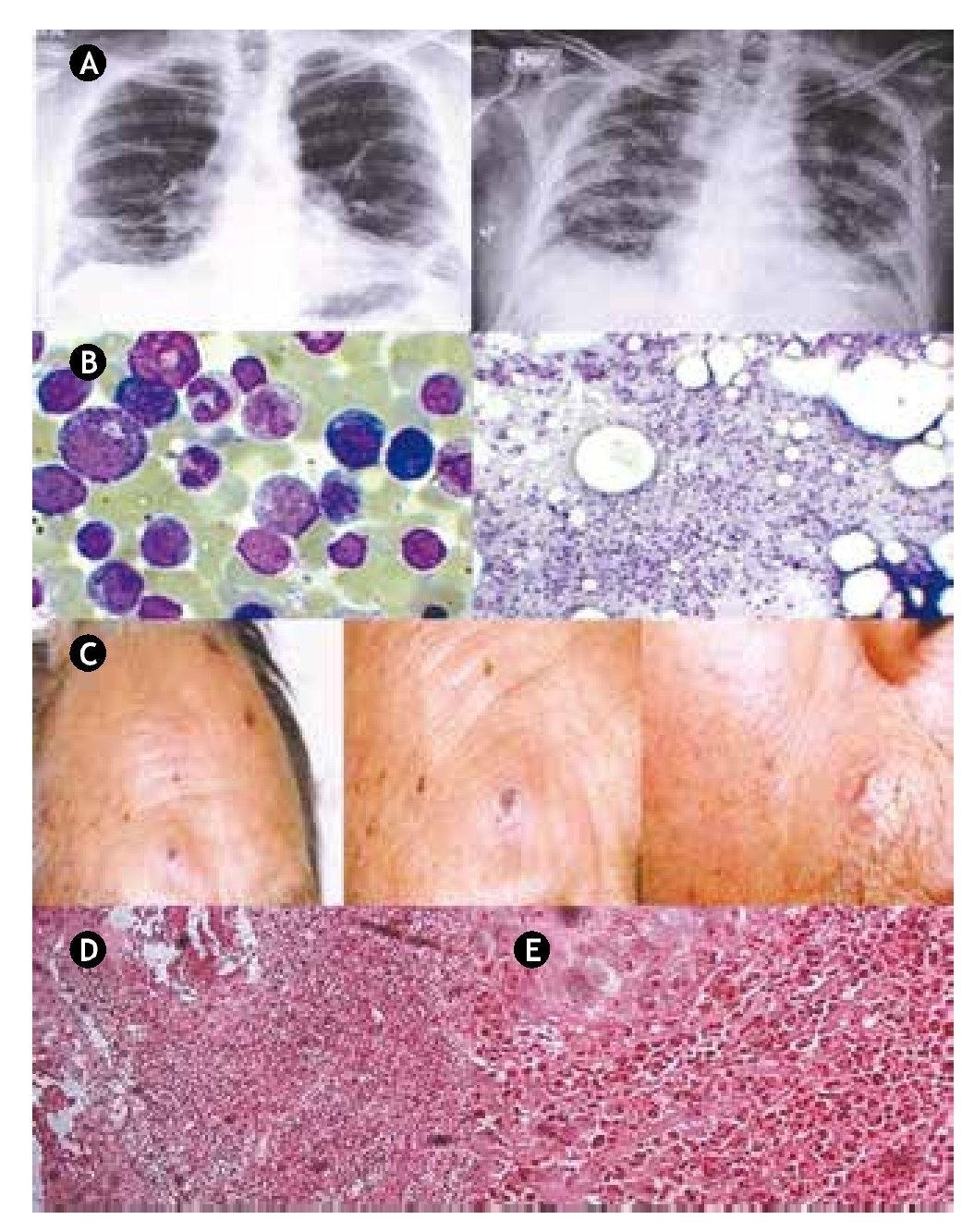
Figure 1 A) The chest X-rays from 2008 (left) show a bibasal reticular infiltrate; 2 years later on the last X-rays (right) diffuse bilateral infiltrates as well as significant progression of pulmonary fibrosis were documented. B) Bone marrow aspirate; moderate granulocytic hyperplasia. C) Sweet syndrome. Sixtyeight male patient with disseminated dermatosis to the face, predominating on the right supraorbital area and near the nasogenian ipsilateral furrow, characterized by ulcerated plaques filled with necrotic central tissue and partly covered with crust, surrounded by eritema, giving a targetoid appearance. D) Microscopic low-power magnification view of a hematoxylin and eosin-stained skin punch biopsy performed on a left cheek lesion, demonstrating slight spongiosis and superficial erosions scattered in the epidermis, with prominent dense infiltration of leucocytes into the dermis. E) Microscopic high-power view demonstrating dense neutrophilic infiltration in the upper and mid-dermis and neutrophil karyorrhexis.
Discussion
Several theories to explain the pathogenesis of Sweet syndrome exist.17 3 are widely supported a type III hypersensitivity reaction, an activation of T cells by antigens or superantigens, and a disturbance in the function of the neutrophiles. An additional theory notes that inappropriate secretion of interleukin 1 (IL-1) can stimulate macrophages and then cause a rise in the production of G-CSF, responsible for increasing IL-8, a chemotactic factor for neutrophils.5,6
Confirming the knowledge about dermatomyositis in that 10% to 25% of the cases progress to a malignancy,18,19 eight years after its diagnosis and successful therapy, the patient developed NHL that was also successfully treated and remained in remission.
Our patient presented three conditions associated with the diagnosis of Sweet syndrome: an hematologic disorder, a severe infectious process, and an autoimmune disease.20 In this case it is difficult to establish definitively whether the syndrome was associated to NHL or its therapy, as his dermatomyositis was not completely controlled, as shown by progressive lung fibrosis and subsequent fatal pneumonia; another possibility is that both conditions acted as a trigger for the development of Sweet syndrome.
Conflicts of interest
The authors have no conflicts of interest to declare.
Funding
No financial support was provided.
Received: November 2013;
Accepted: November 2013
* Corresponding author:
Hematology Departments,
Internal Medicine Division,
"Dr. José Eleuterio González" University Hospital.
Dr. Rodrigo Barragán Building, 2nd Floor.
Madero y Gonzalitos Avenue, Mitras Centro, Z.P.
64460, Monterrey, N. L., Mexico.
Telephone: +52 (81) 1257 2905. Fax: +52 (81) 1257 2906.
E-mail address: carjaime@hotmail.com (J. C. Jaime-Pérez).