



Sturge–Weber syndrome is a congenital vascular disorder characterised by facial capillary malformation (port-wine stain) associated with venous and capillary malformations in the brain and eye. Neurological symptoms and alterations in other locations may also be observed.
ObjectivesThis study describes the clinical and epidemiological characteristics and different treatments in a cohort of patients diagnosed with Sturge–Weber syndrome in a tertiary hospital.
Material and methodsThis comparative, retrospective and cross-sectional study was conducted by reviewing the medical records of patients diagnosed with Sturge–Weber syndrome between 1998 and 2013.
ResultsThe study included 13 patients (54% male, 46% female) diagnosed with Sturge–Weber syndrome. The mean age at diagnosis was 15 months. Leptomeningeal angiomatosis was present in 100% of cases: right hemisphere (46%), left hemisphere (38%), and bilateral (15%). Facial angioma was present in 61% of the cases: right (23%), left (38%) and bilateral (7%). Other skin disorders were found in 23% of the cases, including 2 with hemilateral involvement on the side where facial and leptomeningeal angiomatosis was present and one case of generalised cutis marmorata. Ocular disease was found in 77% of patients; the most common conditions were glaucoma (46%), strabismus (23%) and choroidal angioma (23%). Epilepsy was present in 100% of the cases, with partial seizures (simple or complex) being the most frequent (62%). Seizure control was highly variable; 31% of the patients had needed to try more than 3 drugs, 15% 3 drugs, and 31% 2 drugs, while 23% experienced good seizure control with monotherapy. One patient required surgery for epilepsy (left hemispherectomy) and has been seizure-free since then. The most frequent observations in electroencephalograms were spikes, polyspikes, and wave spikes in the lobes affected by leptomeningeal angiomatosis (46%). Other neurological symptoms were hemiparesis (39%), recurrent headaches (39%), stroke-like episodes (23%), psychomotor retardation (46%), and mental retardation (46%). Leptomeningeal calcifications could be seen in 85% of patient MRIs, as well as increased calcification in 70%; 54% of the patients had been treated with aspirin.
ConclusionsThere are multiple clinical manifestations of Sturge–Weber syndrome. Being familiar with all of them is vitally important for diagnosing and for monitoring and treating the condition correctly, which will improve the quality of life of these patients.
El síndrome de Sturge-Weber es un trastorno vascular congénito caracterizado por una malformación facial capilar (mancha en vino de Oporto) asociada a malformaciones venosas y capilares en el cerebro y en el ojo. También pueden observarse alteraciones en otras localizaciones y síntomas neurológicos.
ObjetivosDescribir las características clínicas y epidemiológicas, así como los diferentes tratamientos realizados en una cohorte de pacientes diagnosticados de síndrome de Sturge-Weber en un hospital terciario.
Material y métodosEstudio comparativo, retrospectivo y transversal, mediante la revisión de historias clínicas de pacientes diagnosticados de síndrome de Sturge-Weber entre los años 1998 y 2013.
ResultadosSe incluyeron 13 pacientes (54% varones, 46% mujeres) diagnosticados de síndrome de Sturge-Weber. La edad media al diagnóstico fue de 15 meses. Presencia de angiomatosis leptomeníngea en el 100% de los casos: hemisferio derecho (46%), hemisferio izquierdo (38%), afectación bilateral (15%). Presencia de angioma facial (61%): derecho (23%), izquierdo (38%) y bilateral (7%). Otras alteraciones cutáneas: 23% de los casos (2 de ellos la afectación en el hemicuerpo del lado en el que se encontraba también la angiomatosis facial y leptomeníngea y en el otro caso la afectación cutánea fue en forma de cutis marmorata generalizada). Encontramos afectación ocular en el 77% de los pacientes, siendo las más frecuentes: glaucoma (46%), estrabismo (23%) y angiomatosis coroidea (23%). Presencia de epilepsia 100% de los casos, siendo las crisis parciales (simples o complejas) las más frecuentes (62%). El control de las crisis epilépticas fue muy variable, ya que el 31% han necesitado probar más de 3 fármacos, 15% 3 fármacos, 31% 2 fármacos y 23% tuvieron buen control con monoterapia. Uno de los pacientes requirió cirugía de la epilepsia (hemisferectomía izquierda), quedando libre de crisis hasta la fecha. En electroencefalogramas lo más frecuente fue: puntas, puntas ondas o polipuntas-ondas en los lóbulos afectados por angiomatosis leptomeníngea (46%). Otros síntomas neurológicos: hemiparesia (39%), cefaleas recurrentes (39%), episodios stroke-like (23%), retraso psicomotor (46%), retraso mental (46%). Presencia calcificaciones leptomeníngeas en la resonancia magnética (85%). Aumento de las calcificaciones en el 70%. Pacientes tratados con ácido acetilsalicílico: 54%.
ConclusionesSon múltiples las manifestaciones clínicas del síndrome de Sturge-Weber, siendo de vital importancia conocerlas todas para poder realizar un correcto diagnóstico, seguimiento y tratamiento de las mismas, mejorando así la calidad de vida de estos pacientes.
Sturge–Weber syndrome (SWS) is a congenital vascular disease characterised by a facial capillary malformation (port-wine stain) associated with venous and capillary malformations in the brain and eyes. Alterations at other locations, such as the buccal cavity or the respiratory tract, can also be observed.1
This congenital disease is sporadic rather than hereditary, although some familial cases have been described.2 Recently, this syndrome has been associated with a mutation in the gene GNAQ.3 Its incidence is between 1/50000 and 1/230000 live births.4 Aetiology of the disease remains unknown, although it has been hypothesised that capillary malformations are the result of somatic mutations in the ectoderm.2
Clinical manifestations of this syndrome are numerous, and a thorough knowledge of all of them is essential to determine the correct diagnosis, follow-up, and treatment to improve patients’ quality of life.
The aim of this study is to describe the clinical and epidemiological characteristics, as well as the different treatments applied to a cohort of patients diagnosed with SWS at a tertiary hospital.
Material and methodsWe conducted a cross-sectional comparative retrospective study by reviewing the data of patients diagnosed with SWS between 1998 and 2013 in the paediatric neurology department at a tertiary hospital.
Data were collected by reviewing the clinical histories of patients diagnosed with SWS over the last 15 years and matching them to the International Classification of Diseases (ICD-10) criteria.
We excluded patients with facial angioma but no confirmed intracranial lesion.
Data obtained from each patient's clinical history were as follows:
- -
Epidemiological variables: sex and age at symptom onset.
- -
Variables related to skin lesions: presence or absence of facial angioma, side affected by the lesion, and skin lesions in other locations.
- -
Variables related to intracranial lesions: presence of leptomeningeal angiomatosis, extension and location of the lesion, affected side.
- -
Variables related to ophthalmological symptoms: presence or absence of eye involvement, type of eye disorder, and treatment used.
- -
Variables related to neurological symptoms: presence or absence of epilepsy, age at onset, symptoms of epileptic seizures, presence or absence of hemiparesis, presence of psychomotor retardation, associated mental retardation, recurrent headache attacks, stroke-like episodes.
- -
Variables related to complementary tests and the results obtained: neurophysiological studies, neuroimaging studies (computed tomography and/or brain magnetic resonance imaging).
- -
Variables related to antiepileptic treatment: antiepileptic drugs, epilepsy surgery.
- -
Variables related to other treatments: antiplatelet drugs, epilepsy surgery.
We included 13 patients (54% males and 46% females) diagnosed with SWS at our hospital between 1998 and 2013.
Age at diagnosis of SWS ranged from one month to 27 months, with a mean age of 15 months.
Leptomeningeal angiomatosis was present in all cases, and it was most frequently located in the right hemisphere (46%) than in the left hemisphere (38%). Two patients displayed bilateral involvement (15%).
In our series, the lesion most frequently identified was leptomeningeal angiomatosis with involvement of more than 3 lobes ipsilaterally or even contralaterally (38.5%), followed by involvement of 3 lobes (31%). Lesions predominantly affected the parietal, temporal, and occipital lobes. We found involvement of only 2 lobes in 23% of the cases (predominantly in the occipitotemporal lobes) and only one patient with involvement of the occipital lobe exclusively.
Facial angioma was not present in all patients; 4 of them (30%) showed no angioma at any location. Of the patients with facial angioma (61%), 3 displayed right facial angioma (23%), 5 on the left side (38%), and only one showed bilateral facial angioma. According to the literature, facial angiomatosis most frequently affects the first branch of the trigeminal nerve; however, it was difficult to delimit the angiomatosis lesion in this group, and this variable was therefore not included in our study.
In this series, skin lesions at locations other than the face were present in 3 of the 13 patients (23%). Two of them were ipsilateral to facial and leptomeningeal angiomatosis; in the third case, skin lesion appeared as generalised cutis marmorata.
Ocular involvement was identified in 77% of the patients, with glaucoma being the most frequent (46%), followed by strabismus (23%), and choroid angiomatosis (23%). Other ocular signs found, although less frequently, were visual field alterations, ophthalmoparesis, and refractive errors. Four patients (30%) presented different eye defects throughout the course of the disease.
Regarding ophthalmological treatment, 61.5% of the patients needed surgery (to treat glaucoma in most cases); the remaining 38.5% of the patients received conservative treatment.
Of the other neurological symptoms typical of this disease, the most frequent was epilepsy, present in 100% of the cases. Partial seizures (simple or complex) were the most frequent (62% of the cases), followed by secondarily generalised partial seizures (31%), and generalised seizures (8%). Age of onset of the first seizure ranged from 2 months to 11 years, with a mean of 26 months. Eight children (62%) presented their first seizure before the age of 12 months, and 75% of them experienced the first seizure at 6 months of age or younger; the most repeated value was 4 months.
Control of epileptic seizures has been very heterogeneous in this group of patients, since 31% of the patients needed more than 3 different drugs to control epilepsy, 15% tried 3 different drugs, 31% tried 2 different drugs, and only 23% of the patients achieved good seizure control with monotherapy.
The most widely used antiepileptic drugs were oxcarbazepine, valproic acid, levetiracetam, and topiramate, with valproic acid used in 92% of the patients, followed by equal percentages of patients treated with oxcarbazepine and levetiracetam (46%). The most widely used drug combinations were valproic acid and levetiracetam, and valproic acid with oxcarbazepine.
In light of one patient's refractory epileptic seizures and symptoms of incipient cognitive impairment, we decided to treat epilepsy with a left hemispherectomy. The patient remains seizure-free to date. The resected section was analysed by the anatomical pathology service. Architecture seemed preserved at the cortical level, with no clearly diminished neuronal component. We observed a mild gliosis and scattered microcalcifications at both the cortical and white matter levels, in addition to occasional perivascular distribution.
The most frequent findings from conventional electroencephalography (EEG) and video-EEG were spikes, spike-waves, or polyspike-wave discharges in the lobes affected by leptomeningeal angiomatosis; these features were present in 46% of the cases. Other electrical manifestations included low-voltage areas (31%), low-voltage-associated spike-waves (15.5%), and areas of slow wave activity (8%).
Other neurological symptoms of SWS detected in our series included hemiparesis, manifesting in 39% of these patients; recurrent headache attacks, in 39% of the patients; and stroke-like episodes in 23%.
We should not overlook associated psychomotor and mental retardation as an additional neurological finding in this syndrome; it is not clear whether they are caused by the disease itself or secondary to epileptic seizures. In our series, 54% of the patients did not develop symptoms of psychomotor or mental retardation. Among the 46% with symptoms, we observed that those presenting signs of psychomotor retardation in their first months would later have intelligence quotients of less than 70 according to psychometric tests.
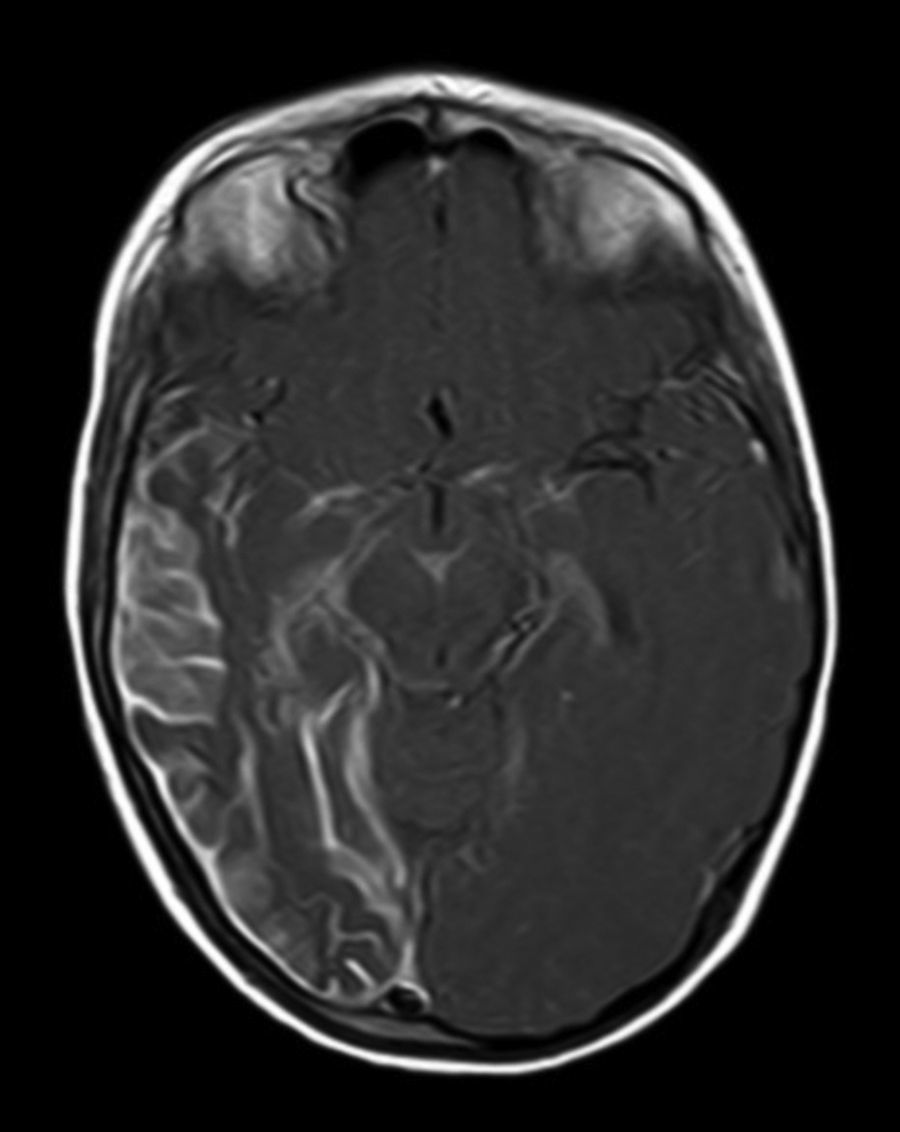
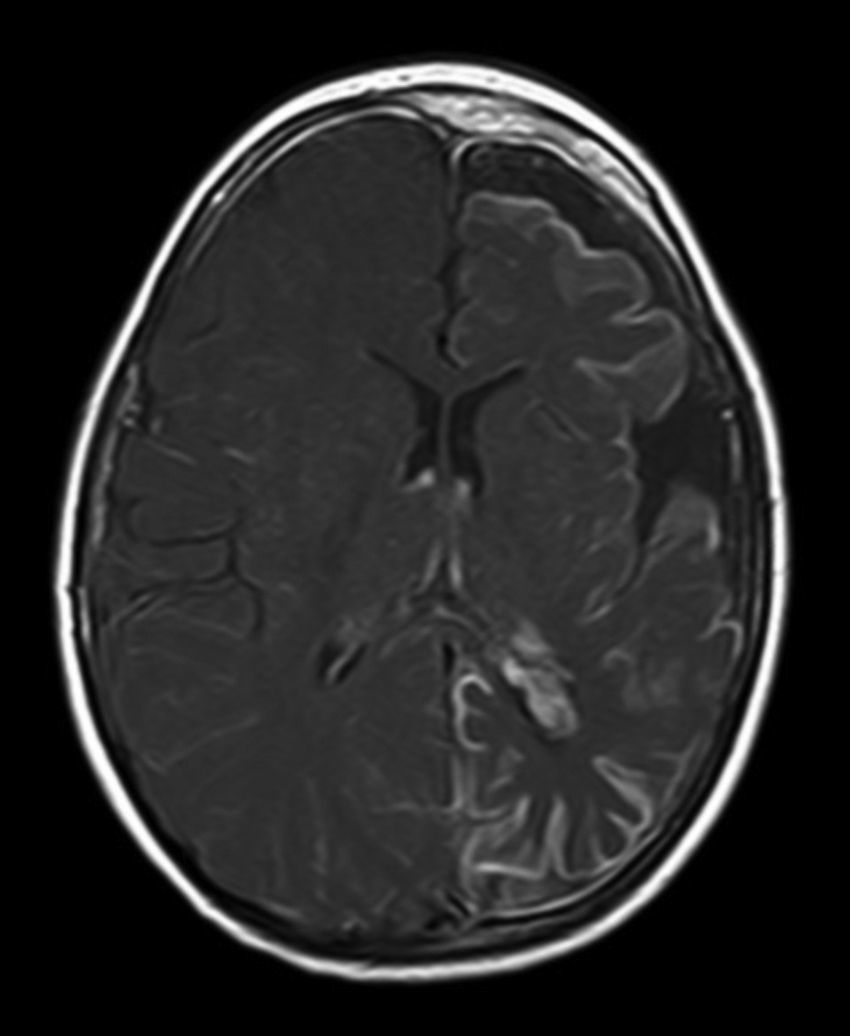
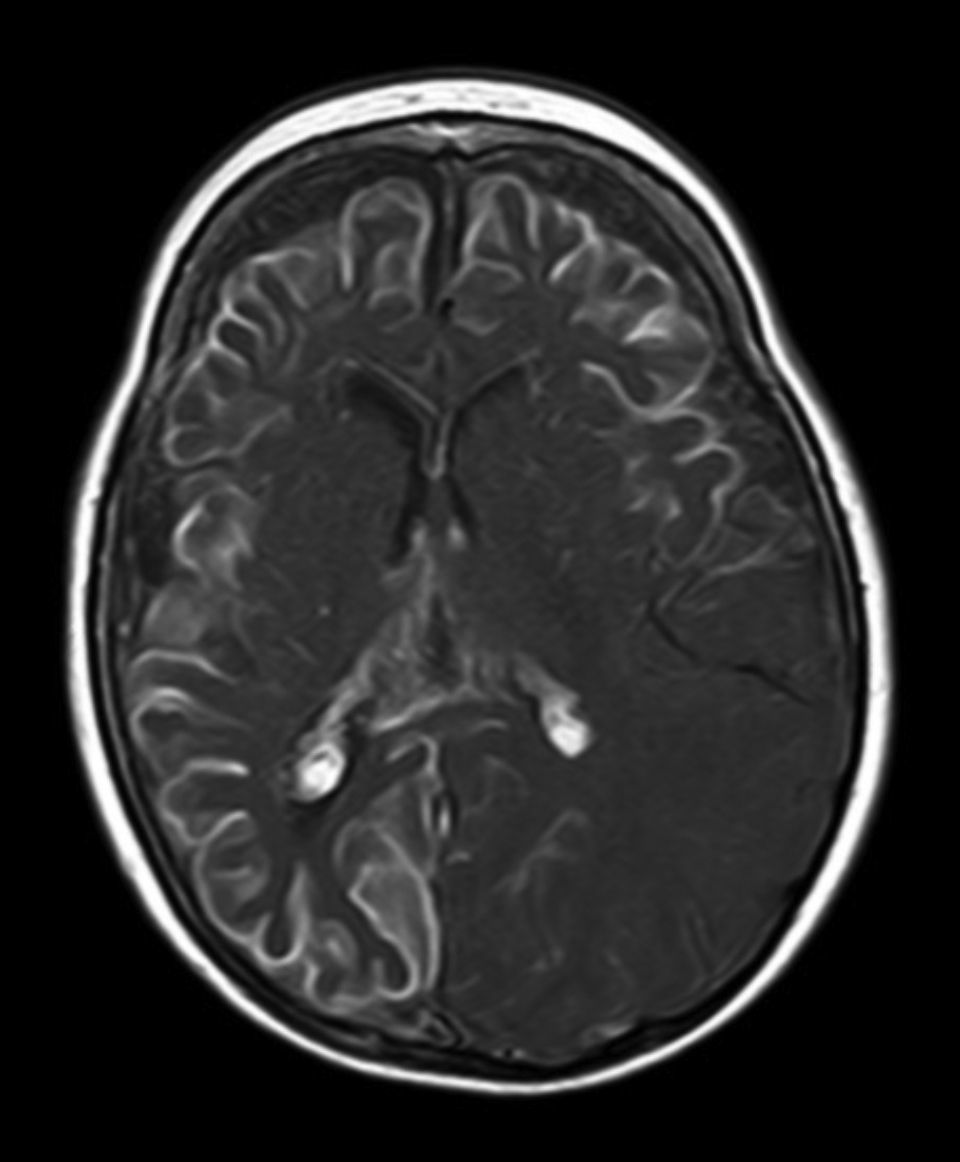
All patients had at least one follow-up magnetic resonance imaging scan performed at some point in time; scans revealed leptomeningeal calcifications in 85% of the patients, with all calcifications located in the angiomatosis area. Subsequent imaging studies performed on these same patients showed increases in the number of calcifications in 70% of them. Other imaging findings in this group of patients included a case of hemimegalencephaly and polymicrogyria, another with secondary atrophy of the right hemipons and hemi-midbrain with opercular syndrome, and 3 patients with atrophy (generalised cortical-subcortical, perirolandic frontotemporal, and left temporal) (Figs. 1 and 2).


Regarding use of coadjuvant treatments, several studies have shown that an antiplatelet drug, acetylsalicylic acid, is effective for decreasing thrombotic and stroke-like episodes in patients with SWS. Fifty-four per cent of the patients in our series were treated with acetylsalicylic acid.
DiscussionSWS was first mentioned in 1860 by Schirmer, a German ophthalmologist, but it was not fully described until 1879 by Sturge. Forty-three years later, in 1922, Weber completed the list of signs by describing the radiological findings.5
The manifestations of this syndrome are multiple; one of its salient features is the port-wine stain or facial angiomatosis, which is the most frequent vascular malformation. It manifests in 0.3% of newborns, but only a small percentage of children with haemangiomas also have SWS. Facial angiomatosis is typically located on the eyelid and the forehead, especially in the distribution of the first and second branch of the trigeminal nerve.
In newborns, the lesion is flat and pink-coloured. As the child ages, the colour may deepen to dark red, and vascular ectasia lesions within the lesion may cause superficial nodularity and blebs. Facial angioma is not present in all patients with SWS6,7; 30% of the patients in our series had no angiomas at any location. In contrast, rather than affecting only one side of the face, skin lesions may occasionally present bilaterally, or even extend to the chest and the limbs.8 Our study includes 2 patients with angiomatosis on one side of the body (ipsilateral to the facial and leptomeningeal lesions) and one patient with a skin lesion manifesting as generalised cutis marmorata.
Several studies have demonstrated that pulsed laser light is effective for treating this type of lesions.9 However, due to the high recurrence rate caused by vessel reperfusion, researchers are investigating new techniques, such as the combination of topical rapamycin and pulsed laser light which is providing good results.10 Cases of intraoral angiomatosis have also been reported. It is present in up to 40% of the cases and occasionally causes significant periodontal alterations that increase the risk of bleeding during dental procedures,11 and even during difficult intubations.12
Leptomeningeal angiomatosis manifests in 10% to 20% of the cases with typical facial angioma, generally on the same side. All patients diagnosed with SWS at our hospital presented leptomeningeal angiomatosis, with or without facial involvement. Parietal and occipital regions are the most frequently affected, although any area of the brain may be involved as our case series illustrates. The occipital lobe was the most frequently affected lobe, whether alone or associated with angiomatosis in other lobes (involvement of more than 3 lobes: 38.5%; 3 lobes: 31%; 2 lobes: 23%; and one lobe: 7.7%). Regarding the side affected by the leptomeningeal angioma, no significant differences have been observed between the 2 hemispheres. Bilateral involvement was detected in 15% of the patients (Fig. 3). Intracranial venous malformations may present in the absence of facial angioma (atypical SWS or type-III SWS), as occurs in 30% of our patients.6,13 Several hypotheses postulate that the angioma present in classic SWS is the result of insufficient retraction and limited maturation of the cephalic venous plexus during the first 3 months of foetal development.13 Other evidence suggests that angiomas are not static; rather, these lesions undergo angiogenic remodelling. These findings may lead to the development of new therapeutic strategies for SWS.14

Ocular signs are present in a large number of patients. They usually manifest as visual field defects, generally as homonymous hemianopsia, due to the presence of angiomas affecting both occipital lobes or optic pathways. The risk of glaucoma is higher in patients younger than 10, and it occurs in 30% to 70% of those diagnosed with SWS.15,16 Continuous follow-up was necessary, even for patients whose intraocular pressure was initially normal.
Other ophthalmological complications include choroidal haemangiomas, vascular malformations in the episcleral lamina or conjunctiva, and heterochromia.
In our series, ocular involvement was identified in 77% of the patients; glaucoma (46%) was the most frequent as reported in the literature, followed by strabismus (23%), and choroid angiomatosis (23%). Only one patient displayed hemianopsia with no other eye disorder. Four patients (30%) presented different eye disorders as the condition progressed. Several articles address the different treatments for eye disorders in SWS: trabeculectomy for glaucoma, photodynamic therapy, etc.15,17 Only 61.5% of the patients in our series needed surgery (mostly to treat glaucoma); the other 38.5% of the patients received conservative treatment.
Epilepsy is the most important of the neurological alterations potentially associated with this syndrome. Seizures are frequently the first symptom of SWS, and they manifest in 75% to 80% of these patients. While seizures may occur at any age, onset usually occurs in early childhood.18 The initial seizures are typically focal, but they often evolve to become generalised tonic-clonic seizures. Infantile spasms, myoclonic seizures, or atonic seizures may also be present. In our study, epilepsy manifested in 100% of the cases. Partial seizures (simple or complex) were the most frequent (62% of the cases), followed by secondarily generalised partial seizures (31%) and generalised seizures (8%). Age at seizure onset ranged from 2 months to 11 years, with a mean age of 26 months. Eight children (62%) presented their first seizure before the age of 12 months, and 75% of them suffered the first seizure at 6 months of age or younger.
The degree of seizure control achieved with antiepileptics was variable and unpredictable. Some patients experience long seizure-free periods, even without medication, whereas others suffer frequent or prolonged attacks despite taking high doses of various antiepileptics. Seizure control is very heterogeneous in this group of patients, considering that 31% of the patients needed more than 3 different drugs to control epilepsy, 15% tried 3 drugs, 31% tried 2 drugs, and only 23% of the patients achieved good control with monotherapy.
Seizure onset, age at symptom onset, and response to treatment all affect prognosis. Seizure onset in children younger than one year and poor response to antiepileptics are associated with a higher probability of cognitive impairment.16
One theory to explain epileptogenesis in patients with SWS-associated epilepsy is chronic ischaemia in cortical areas affected by leptomeningeal angiomatosis. Nevertheless, we have found cases of patients with intractable epilepsy (caused by SWS) whose epileptogenic focus was an area of type-IIa focal cortical dysplasia near the area displaying leptomeningeal angiomatosis.19,20
Several articles have analysed surgical treatment of refractory epilepsy in these patients, and particularly the use of hemispherectomy to achieve better seizure control.21,22 One patient in our study underwent left hemispherectomy to treat refractory epilepsy and symptoms of cognitive impairment; results were excellent since no seizures occurred in the 7 months between the surgery and the date of data collection.
Hemiparesis, present in 39% of the cases analysed, frequently develops in its acute form at the time of seizure onset.21 The paretic limb does not develop normally and becomes atrophied. Affected children sometimes present progressive loss of motor function caused by an unknown mechanism. One theory points to the cumulative effect of repeated thrombotic episodes inside the leptomeningeal malformation, or chronic alteration of blood flow. Numerous studies seem to have demonstrated the efficacy of low doses of acetylsalicylic acid for reducing thrombotic episodes in these patients.23–25
Patients with SWS may also develop stroke-like episodes that cause transient neurological deficits often lasting several days or weeks.18 Of the cases we studied, 23% had presented one or more stroke-like episodes at some point. These episodes may occur spontaneously, accompany a cluster of seizures, or precede a migraine or a seizure. Treatment with low doses of acetylsalicylic acid has also proved effective for reducing these episodes.23–25 Fifty-four percent of children in our study needed treatment with antiplatelet drugs due to thrombotic or stroke-like episodes. Subjective improvement was reported in the form of decreased frequency and severity.
Headaches, predominantly migraine, are also more frequent in the context of this syndrome and they manifest at earlier ages than in the general population.18 Our series reports recurrent headache attacks in 39% of the patients.
Children with SWS usually develop normally in the first few months after birth, but developmental delays will manifest at a later stage.26 Cognitive function is more limited in patients displaying bilateral intracerebral lesions.27 We found that 46% of the children studied in our series presented signs of psychomotor retardation. Subsequent psychometric testing showed total intelligence quotients of less than 70 in these children.
Poorer cognitive function and presence of epilepsy in patients with SWS is also associated with a higher risk of behavioural problems. However, behavioural problems may also occur in patients with normal intelligence.28,29
Other symptoms that may be associated with SWS have been analysed in recent years. For example, researchers have observed that growth hormone deficiency is up to 18 times more frequent in these patients than in the general population. This deficiency occurs with no neuroimaging evidence of pituitary or hypothalamic anomalies.30 Cases of partial hypopituitarism31 and central hypothyroidism that may be related to the use of antiepileptics have also been reported.32 Since effective treatment for these symptoms exists, we recommend testing to facilitate early diagnosis of these patients.
Diagnosis of SWS is based on the presence of facial angioma and leptomeningeal angiomatosis. As mentioned earlier, some patients with intracranial malformations and no facial lesions may also develop neurological problems.
The neuroimaging technique of choice for diagnosis is gadolinium contrast magnetic resonance imaging, since it can reveal leptomeningeal angiomatosis and the extent to which it affects brain structures.33 In some cases, leptomeningeal involvement cannot be detected by neuroimaging techniques during childhood and only becomes apparent later in life.2 All patients underwent at least one magnetic resonance imaging scan at some point; we found leptomeningeal calcifications in 85% of the patients, all of them located in the area affected by angiomatosis. Subsequent imaging studies of the same patients showed increases in the number of calcifications in 70% of them.
When magnetic resonance imaging is not available, a cranial computed tomography may be able to show brain calcifications and provide some anatomical information. Single-photon emission computed tomography has been able to show decreased blood flow in the part of the brain affected by leptomeningeal angiomatosis.18
Patients with SWS may display multiple radiological findings other than those previously mentioned. These include lobe atrophy or atrophy at other locations (in 4 of our patients), choroid plexus hypertrophy, venous flow anomalies, prominent deep collateral venous system, etc.
EEG is a non-invasive method of assessing brain function, which can be repeated as frequently as necessary.18 The most common EEG abnormality is decreased voltage in the affected hemisphere, but this was not the case in our study. Among our patients, the most frequent abnormalities in EEG studies were spikes, spike-waves, or polyspike-waves (46%), followed by low-voltage areas (31%), low-voltage-associated spike-waves (15.5%), and areas of slow wave activity (8%).
Ongoing studies with EEG examine whether this tool may be helpful in achieving better outcomes and selecting treatments.18
Some studies of children with SWS have revealed that Doppler ultrasonography may be a useful non-invasive tool for assessing the severity of anomalies in blood flow and for monitoring changes over time.18
Marler et al. have developed vascular biomarkers (increased metalloproteinases in urine) and described non-invasive vascular profiles of patients with different vascular disorders, including patients with SWS. Continuous identification and validation of biomarkers of this syndrome will be a significant component of future therapeutic approaches.18
There is no specific treatment for SWS. Treatment of cutaneous, ocular, neurological, and endocrine manifestations yields uneven results. Understanding and managing antiepileptic treatment in these children is of vital importance, since some anticonvulsant agents, such as topiramate, may cause acute angle-closure glaucoma and worsen the patient's prognosis.18
There is increasing consensus that low doses of acetylsalicylic acid may be beneficial, based on the idea that antithrombotic treatment may prevent the exacerbation of anomalies in cerebral blood flow and of the hypoxic-ischaemic brain lesion.23–25 Furthermore, there are other strategies for preventing stroke-like episodes, including annual influenza vaccination, maintaining good hydration during disease, and treating fevers, but these have not been thoroughly studied.18
Prognosis in SWS depends on the extension of leptomeningeal angiomatosis and its effect on perfusion in the cerebral cortex, as well as severity of eye impairment. Prognosis also depends on age at seizure onset and on the effectiveness of seizure control. Neurological functions may deteriorate with age. As a result, approximately half of the adults with SWS present impairment, including those who were initially normal.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Maraña Pérez AI, Ruiz-Falcó Rojas ML, Puertas Martín V, Domínguez Carral J, Carreras Sáez I, Duat Rodríguez A, et al. Análisis del síndrome de Sturge-Weber: estudio retrospectivo de múltiples variables asociadas. Neurología. 2017;32:363–370.
