



El síndrome de Sturge-Weber es un trastorno vascular congénito caracterizado por una malformación facial capilar (mancha en vino de Oporto) asociada a malformaciones venosas y capilares en el cerebro y en el ojo. También pueden observarse alteraciones en otras localizaciones y síntomas neurológicos.
ObjetivosDescribir las características clínicas y epidemiológicas, así como los diferentes tratamientos realizados en una cohorte de pacientes diagnosticados de síndrome de Sturge-Weber en un hospital terciario.
Material y métodosEstudio comparativo, retrospectivo y transversal, mediante la revisión de historias clínicas de pacientes diagnosticados de síndrome de Sturge-Weber entre los años 1998 y 2013.
ResultadosSe incluyeron 13 pacientes (54% varones, 46% mujeres) diagnosticados de síndrome de Sturge-Weber. La edad media al diagnóstico fue de 15 meses. Presencia de angiomatosis leptomeníngea en el 100% de los casos: hemisferio derecho (46%), hemisferio izquierdo (38%), afectación bilateral (15%). Presencia de angioma facial (61%): derecho (23%), izquierdo (38%) y bilateral (7%). Otras alteraciones cutáneas: 23% de los casos (2 de ellos la afectación en el hemicuerpo del lado en el que se encontraba también la angiomatosis facial y leptomeníngea y en el otro caso la afectación cutánea fue en forma de cutis marmorata generalizada). Encontramos afectación ocular en el 77% de los pacientes, siendo las más frecuentes: glaucoma (46%), estrabismo (23%) y angiomatosis coroidea (23%). Presencia de epilepsia 100% de los casos, siendo las crisis parciales (simples o complejas) las más frecuentes (62%). El control de las crisis epilépticas fue muy variable, ya que el 31% han necesitado probar más de 3 fármacos, 15% 3 fármacos, 31% 2 fármacos y 23% tuvieron buen control con monoterapia. Uno de los pacientes requirió cirugía de la epilepsia (hemisferectomía izquierda), quedando libre de crisis hasta la fecha. En electroencefalogramas lo más frecuente fue: puntas, puntas ondas o polipuntas-ondas en los lóbulos afectados por angiomatosis leptomeníngea (46%). Otros síntomas neurológicos: hemiparesia (39%), cefaleas recurrentes (39%), episodios stroke-like (23%), retraso psicomotor (46%), retraso mental (46%). Presencia calcificaciones leptomeníngeas en la resonancia magnética (85%). Aumento de las calcificaciones en el 70%. Pacientes tratados con ácido acetilsalicílico: 54%.
ConclusionesSon múltiples las manifestaciones clínicas del síndrome de Sturge-Weber, siendo de vital importancia conocerlas todas para poder realizar un correcto diagnóstico, seguimiento y tratamiento de las mismas, mejorando así la calidad de vida de estos pacientes.
Sturge-Weber syndrome is a congenital vascular disorder characterised by facial capillary malformation (port-wine stain) associated with venous and capillary malformations in the brain and eye. Neurological symptoms and alterations in other locations may also be observed.
ObjectivesThis study describes the clinical and epidemiological characteristics and different treatments in a cohort of patients diagnosed with Sturge-Weber syndrome in a tertiary hospital.
Material and methodsThis comparative, retrospective and cross-sectional study was conducted by reviewing the medical records of patients diagnosed with Sturge-Weber syndrome between 1998 and 2013.
ResultsThe study included 13 patients (54% male, 46% female) diagnosed with Sturge-Weber syndrome. The mean age at diagnosis was 15 months. Leptomeningeal angiomatosis was present in 100% of cases: right hemisphere (46%), left hemisphere (38%), and bilateral (15%). Facial angioma was present in 61% of the cases: right (23%), left (38%) and bilateral (7%). Other skin disorders were found in 23% of the cases, including 2 with hemilateral involvement on the side where facial and leptomeningeal angiomatosis was present and one case of generalised cutis marmorata. Ocular disease was found in 77% of patients; the most common conditions were glaucoma (46%), strabismus (23%) and choroidal angioma (23%). Epilepsy was present in 100% of the cases, with partial seizures (simple or complex) being the most frequent (62%). Seizure control was highly variable; 31% of the patients had needed to try more than 3 drugs, 15% 3 drugs, and 31% 2 drugs, while 23% experienced good seizure control with monotherapy. One patient required surgery for epilepsy (left hemispherectomy) and has been seizure-free since then. The most frequent observations in electroencephalograms were spikes, polyspikes, and wave spikes in the lobes affected by leptomeningeal angiomatosis (46%). Other neurological symptoms were hemiparesis (39%), recurrent headaches (39%), stroke-like episodes (23%), psychomotor retardation (46%), and mental retardation (46%). Leptomeningeal calcifications could be seen in 85% of patient MRIs, as well as increased calcification in 70%; 54% of the patients had been treated with aspirin.
ConclusionsThere are multiple clinical manifestations of Sturge-Weber syndrome. Being familiar with all of them is vitally important for diagnosing and for monitoring and treating the condition correctly, which will improve the quality of life of these patients.
El síndrome de Sturge-Weber es un trastorno vascular congénito caracterizado por una malformación facial capilar (mancha en vino de Oporto) asociada a malformaciones venosas y capilares en el cerebro y en el ojo, aunque también pueden observarse alteraciones en otras localizaciones, como pueden ser la cavidad oral o la vía respiratoria1.
Es un trastorno congénito y esporádico de carácter no hereditario, aunque se han descrito algunos casos familiares2. Recientemente se ha descrito la asociación de este síndrome con una mutación en el gen GNAQ3. Tiene una incidencia de 1/50.000-1/230.000 recién nacidos vivos4. La etiología es desconocida, aunque se baraja la hipótesis de que las malformaciones capilares son el resultado de mutaciones somáticas en los tejidos fetales ectodérmicos2.
Son múltiples las manifestaciones clínicas de este síndrome, siendo de vital importancia conocerlas todas para poder realizar un correcto diagnóstico, seguimiento y tratamiento de las mismas, mejorando así la calidad de vida de estos pacientes.
Los objetivos de este trabajo son describir las características clínicas y epidemiológicas, así como los diferentes tratamientos realizados en una cohorte de pacientes diagnosticados de síndrome de Sturge-Weber en un hospital terciario.
Material y métodosSe ha realizado un estudio comparativo, retrospectivo y transversal, mediante la revisión de datos de pacientes diagnosticados de síndrome de Sturge-Weber entre los años 1998 y 2013 en las consultas de neuropediatría de un hospital de tercer nivel.
La recogida de datos se realizó mediante la revisión de las historias clínicas a través del sistema de codificación de pacientes (CIE-10) diagnosticados de síndrome de Sturge-Weber durante los últimos 15 años.
Se han excluido del estudio aquellos pacientes con angioma facial sin confirmación de lesión intracraneal.
Los datos que se han obtenido de la historia clínica de cada paciente han sido:
- -
Variables epidemiológicas: sexo y edad en el momento del inicio de la clínica.
- -
Variables relacionadas con las lesiones cutáneas: presencia o ausencia de angioma facial, lateralidad del mismo y lesiones cutáneas en otras localizaciones.
- -
Variables relacionadas con las lesiones intracraneales: presencia de angiomatosis leptomeníngea, extensión y localización de la lesión, lateralidad.
- -
Variables relacionadas con la clínica oftalmológica: presencia o ausencia de alteración ocular, tipo de afectación oftalmológica, tratamiento empleado.
- -
Variables relacionadas con síntomas neurológicos: presencia o ausencia de epilepsia, edad de inicio y semiología de las crisis epilépticas, presencia o ausencia de hemiparesia, aparición de retraso psicomotor, retraso mental asociado, presencia de cefaleas recurrentes, episodios stroke-like.
- -
Variables relacionadas con las exploraciones complementarias realizadas y la obtención de resultados en las mismas: estudios neurofisiológicos, estudios de neuroimagen (tomografía computarizada y/o resonancia magnética cerebral).
- -
Variables relacionadas con el tratamiento antiepiléptico: fármacos antiepilépticos recibidos, realización de cirugía de la epilepsia.
- -
Variables relacionadas con otros tratamientos recibidos: tratamiento antiagregante, cirugía de la epilepsia.
Se incluyeron en el estudio 13 pacientes (54% varones y 46% mujeres) diagnosticados de síndrome de Sturge-Weber en nuestro hospital entre los años 1998 y 2013.
La edad media al diagnóstico de síndrome de Sturge-Weber osciló entre el mes de vida y los 27 meses, con una media de edad de 15 meses.
En todos ellos estuvo presente la angiomatosis leptomeníngea, siendo la localización más frecuente en el hemisferio derecho con un 46%, frente a un 38% de afectación hemisférica izquierda. Cabe mencionar que en 2 de los pacientes la afectación fue bilateral (15%).
En nuestra serie lo más frecuente encontrado fue la angiomatosis leptomeníngea, con afectación de más de 3 lóbulos (38,5%), siendo ipsilaterales o incluso contralaterales, seguido de afectación de 3 lóbulos (31%), con predominio de afectación parieto-temporooccipital. Encontramos afectación de solo 2 lóbulos en el 23% de los casos (predominio lóbulos temporooccipitales) y solo en un paciente estuvo afecto el lóbulo occipital únicamente.
La presencia de angioma facial no fue constante en todos los pacientes, ya que 4 de ellos (30%) no presentaron angioma en ninguna localización. De los pacientes que sí presentaron angioma facial (61%) 3 de ellos tuvieron angioma facial derecho (23%), 5 en hemicara izquierda (38%) y solo en uno de ellos fue bilateral. Según la literatura la afectación de la angiomatosis facial es más frecuente en la primera rama del nervio trigémino; no obstante, en el grupo de pacientes de este estudio fue difícil delimitar la angiomatosis, motivo por el que esta variable no se ha incluido en el estudio.
En esta serie se encontraron alteraciones cutáneas en otras localizaciones (distintas de la afectación facial) en 3 de los 13 pacientes (23%), siendo en 2 de ellos la afectación en el hemicuerpo del lado en el que se encontraba también la angiomatosis facial y leptomeníngea, y en el otro caso la afectación cutánea fue en forma de cutis marmorata generalizada.
La afectación ocular se produjo en el 77% de los pacientes, siendo la enfermedad más frecuente el glaucoma (46%), seguido del estrabismo (23%) y la angiomatosis coroidea (23%). Otras de las alteraciones oculares encontradas, aunque con menor frecuencia, fueron defectos campimétricos, oftalmoparesia y defectos de refracción. Cuatro de los pacientes (30%) sufrieron varios defectos oculares a lo largo de su evolución.
Respecto al tratamiento oftalmológico empleado el 61,5% de los pacientes precisó intervenciones quirúrgicas (mayoritariamente para el tratamiento del glaucoma) frente al 38,5% de los pacientes en los que se hizo tratamiento conservador.
Entre las otras manifestaciones neurológicas propias de la enfermedad el síntoma más frecuente fue la epilepsia en el 100% de los casos, siendo las crisis parciales (simples o complejas) las más frecuentes en el 62% de los casos, seguido de las crisis parciales secundariamente generalizadas (31%) y las crisis generalizadas (8%). El intervalo de edad de la aparición de la primera crisis oscila entre los 2 meses de vida y los 11 años, con una media de edad de 2 años y 2 meses. El 62% (8 niños) tuvieron la primera crisis con menos de 12 meses, y de estos, el 75% sufrieron la primera crisis con 6 meses o menos, siendo el valor más repetido los 4 meses.
El control de las crisis epilépticas ha sido muy variable en este grupo de pacientes, ya que hasta el 31% de los pacientes han necesitado probar más de 3 fármacos para el manejo de la epilepsia, el 15% han probado solo 3 fármacos, el 31% han necesitado utilizar 2 fármacos y solo el 23% de los pacientes han estado bien controlados con monoterapia.
Los fármacos antiepilépticos más empleados fueron la oxcarbacepina, el ácido valproico, el levetiracetam y el topiramato, siendo el ácido valproico el fármaco más utilizado en el 92% de los pacientes, seguido de la oxcarbacepina y el levetiracetam con igual frecuencia (46%). Las combinaciones de fármacos más manejadas fueron ácido valproico y levetiracetam, y ácido valproico junto a oxcarbacepina.
En uno de los pacientes, ante la refractariedad de las crisis epilépticas y el inicio de síntomas de deterioro cognitivo, se decidió realizar cirugía de la epilepsia consistente en una hemisferectomía izquierda, quedando el paciente libre de crisis hasta la fecha. La pieza resecada se analizó por el servicio de anatomía patológica, visualizándose al nivel de la corteza cerebral una arquitectura conservada, sin clara disminución en el componente neuronal, apreciándose gliosis leve y microcalcificaciones que aparecieron de forma salpicada, tanto a nivel de la corteza como en la sustancia blanca, con ocasional distribución perivascular de las mismas.
Entre los hallazgos encontrados en los electroencefalogramas convencionales y vídeo-electroencefalogramas lo más frecuente fue la aparición de puntas, puntas ondas o polipuntas-ondas en los lóbulos afectados por angiomatosis leptomeníngea en el 46% de los casos. Otras manifestaciones eléctricas encontradas fueron zonas de hipovoltaje (31%), puntas ondas asociadas a hipovoltaje (15,5%) y regiones de lentificación (8%).
Otros de los síntomas neurológicos presentes en el síndrome de Sturge-Weber que encontramos en nuestra serie fue la hemiparesia, que se presentó en el 39% de los pacientes; las cefaleas recurrentes, que se hallaron también en el 39% de los niños estudiados, y los episodios stroke-like que aparecieron en el 23%.
No debemos olvidar la presencia en este síndrome de retraso psicomotor y retraso mental asociado como otro de los hallazgos neurológicos encontrados, en los que no está bien definido si son propios de la enfermedad o secundarios a las crisis epilépticas. En nuestra serie encontramos que el 54% de los pacientes no presentaron signos de retraso psicomotor ni retraso mental, frente al 46% en los que sí estuvieron presentes, habiendo una relación directa entre los pacientes que en los primeros meses de la vida presentaron signos de retraso psicomotor y en los que posteriormente se pudo objetivar mediante pruebas psicométricas un cociente intelectual por debajo de 70.
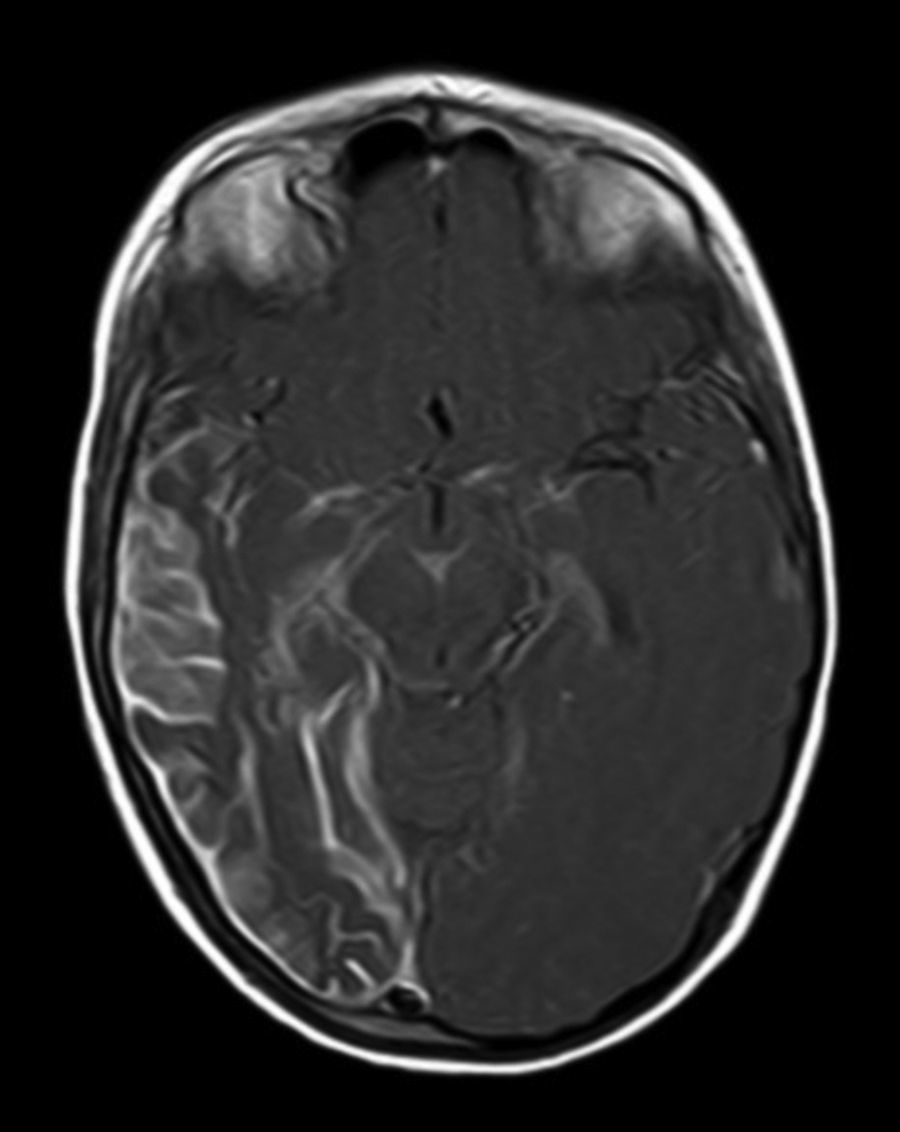
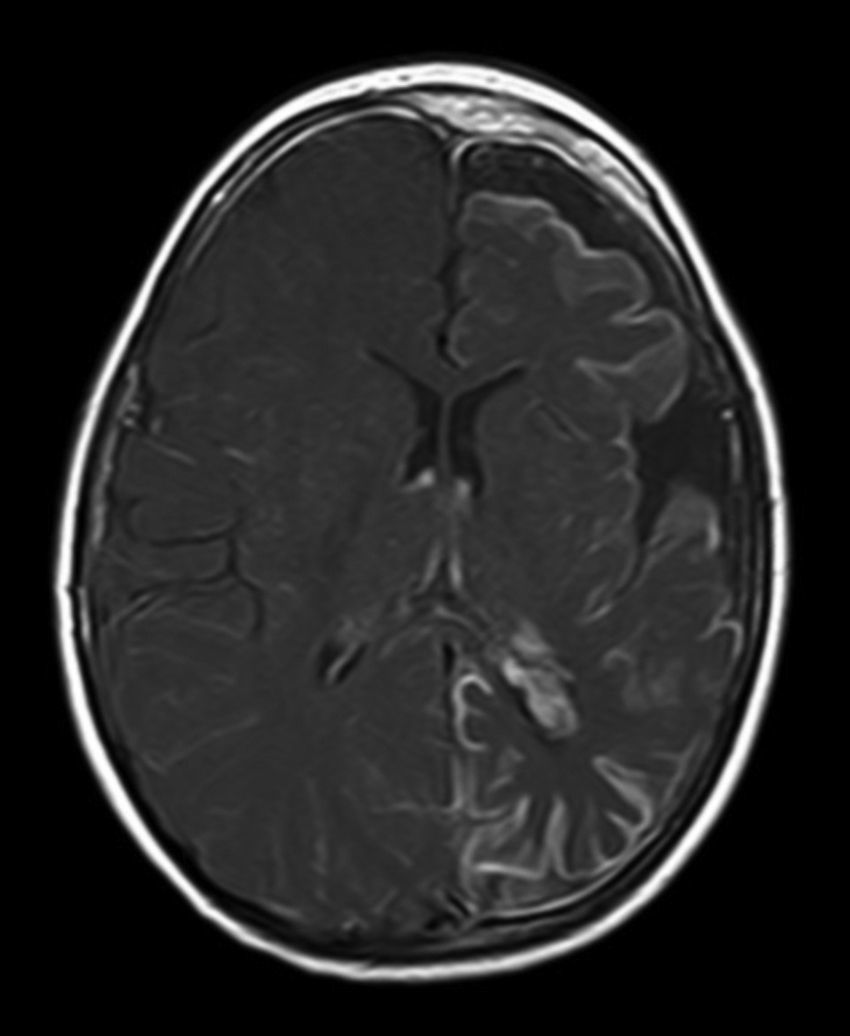
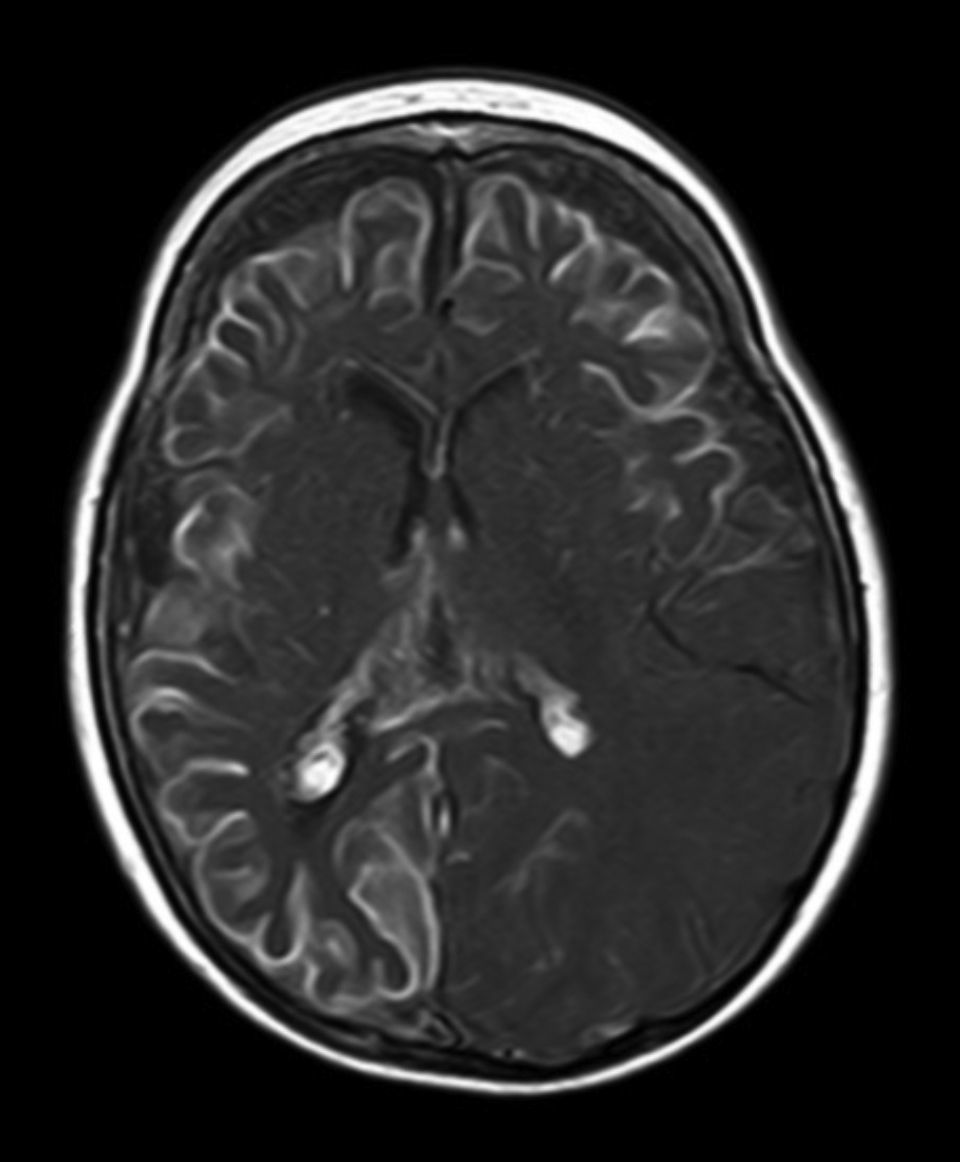
A todos los pacientes se les realizó al menos una resonancia magnética de control a lo largo de su evolución, encontrándose calcificaciones leptomeníngeas en el 85% de los pacientes, todas ellas localizadas en el territorio de la angiomatosis. En controles radiológicos posteriores realizados a estos mismos pacientes se visualizó el aumento de las calcificaciones en el 70% de los mismos. Otros hallazgos radiológicos encontrados en este grupo de pacientes fueron: un paciente con hemimegalencefalia y polimicrogiria, otro con atrofia secundaria en hemiprotuberancia, hemimesencéfalo derecho y displasia opercular y 3 pacientes con atrofia (una córtico-subcortical generalizada, otra frontoparietal perirrolándica y otra temporal izquierda) (figs. 1 y 2).


Con respecto a la utilización de otros tratamientos coadyuvantes, diversos han sido los estudios que reportan la eficacia del tratamiento con un antiagregante, el ácido acetilsalicílico, en pacientes con síndrome de Sturge-Weber, disminuyendo los episodios trombóticos y stroke-like. En nuestra serie de pacientes encontramos que el 54% estaba tratado con el ácido acetilsalicílico, frente al 46% que no lo tomaba.
DiscusiónEl síndrome de Sturge-Weber fue descrito por primera vez en 1860 por Schirmer, oftalmólogo alemán, pero no se describió de forma completa hasta 1879 por Sturge. Cuarenta y tres años más tarde, en 1922, Weber completó el cuadro con la descripción de las alteraciones radiológicas5.
Múltiples son las manifestaciones encontradas en este síndrome, entre ellas destacan la aparición de «mancha en vino de Oporto» o angiomatosis facial, que es el tipo más común de malformación vascular. Ocurre hasta en el 0,3% de los recién nacidos, sin embargo, solo una pequeña proporción de niños con hemangiomas tienen síndrome de Sturge-Weber. Generalmente se encuentra localizada en el párpado superior y la frente, especialmente en la distribución de la primera y segunda rama del nervio trigémino.
En el recién nacido la lesión es plana, de color rosa, y con la edad suele oscurecerse hasta un rojo intenso, produciéndose ectasias vasculares en su interior que originan nodularidad superficial y ampollas. La presencia de angioma facial no es constante en todos los pacientes con síndrome de Sturge-Weber6,7, como así ha ocurrido en el 30% de los pacientes de nuestra serie, que no han presentado angioma en ninguna localización. En contraposición, en otras ocasiones la lesión cutánea no solo afecta una parte de la cara, sino que puede extenderse a ambos lados de la misma, así como al tronco y a las extremidades8. En nuestro estudio hemos encontrado 2 pacientes con angiomatosis en un hemicuerpo (ipsilateral a la lesión facial y leptomeníngea) y en otro de los casos la afectación cutánea se produjo en forma de cutis marmorata generalizado.
Varios estudios han demostrado la eficacia del tratamiento con láser pulsado en este tipo de lesiones9, aunque dada la alta frecuencia de recurrencias debido a la reperfusión de vasos sanguíneos, se está investigando sobre la utilización de nuevas técnicas, como por ejemplo la asociación de rapamicina tópica junto con el láser pulsado con buenos resultados10. También se han reportado casos de angiomatosis intraoral, presente hasta en el 40% de los casos, provocando en ocasiones importantes alteraciones periodontales, lo que aumenta el riesgo de sangrado durante los procedimientos dentales11, e incluso intubaciones complicadas12.
La angiomatosis leptomeníngea ocurre entre el 10-20% de los casos en los que aparece un angioma facial típico, generalmente en el mismo lado. El 100% de los pacientes diagnosticados de síndrome de Sturge-Weber en nuestro centro presentaban angiomatosis leptomeníngea (con o sin afectación facial). Se afectan más comúnmente las regiones parietales y occipitales, aunque cualquier parte del cerebro puede verse implicada, como se demuestra en nuestra serie de casos. El lóbulo más frecuentemente afectado ha sido el lóbulo occipital, con mayor prevalencia asociado también a angiomatosis en otros lóbulos (afectación de más 3 lóbulos: 38,5%, 3 lóbulos: 31%, 2 lóbulos: 23% y un lóbulo: 7,7%). Con respecto a la lateralidad del angioma leptomeníngeo no ha habido diferencias significativas entre ambos hemisferios, encontrando también afectación bilateral en un 15% de los pacientes (fig. 3). En ocasiones la malformación intracraneal puede existir sin acompañarse de angioma facial (SWS atípico o SWS tipo iii), como ha ocurrido en el 30% de nuestros pacientes6,13. Existen hipótesis de que el angioma encontrado en el síndrome Sturge-Weber clásico es el resultado de la insuficiente retracción y escasa maduración del plexo venoso cefálico en el primer trimestre del desarrollo13. Así mismo, también se han encontrado pruebas de que los angiomas no son lesiones estáticas, sino más bien se someten a remodelado angiogénico. Estos hallazgos podrían conducir al desarrollo de nuevas estrategias terapéuticas en el síndrome de Sturge Weber14.

Las alteraciones oculares ocurren en un alto número de pacientes. En ocasiones se presentan como defectos del campo visual, por lo general una hemianopsia homónima, debido a la presencia de angiomas afectando ambos lóbulos occipitales o las vías ópticas. El glaucoma, cuyo riesgo es mayor en la primera década de la vida, se produce entre el 30-70% de los pacientes afectos de síndrome de Sturge-Weber15,16. Se necesita una vigilancia continua, incluso en pacientes con presión intraocular inicialmente normal.
Otras complicaciones oftalmológicas incluyen hemangiomas coroideos, malformaciones vasculares a nivel epiescleral o conjuntival y heterocromía del iris.
En nuestra serie la enfermedad ocular se produjo en el 77% de los casos, siendo el glaucoma (46%) la manifestación más frecuente (como se reporta en la literatura), seguido del estrabismo (23%) y de la angiomatosis coroidea (23%). Solo uno de los pacientes presentó hemianopsia sin otra alteración ocular. Cuatro de los pacientes (30%) sufrieron varias afectaciones oculares a lo largo de su evolución. Múltiples son los artículos que abordan los diferentes tratamientos de alteraciones oculares en el síndrome de Stuge-Weber: trabeculectomía para el glaucoma, terapia fotodinámica, etc.15,17. En nuestra serie de casos solo el 61,5% de los pacientes precisaron intervenciones quirúrgicas (mayoritariamente para el tratamiento del glaucoma) frente al 38,5% de los pacientes en los que se hizo tratamiento conservador.
Entre las alteraciones neurológicas que pueden aparecer destaca la presencia de epilepsia. Las crisis son a menudo el primer síntoma de SWS, desarrollándose en el 75-80% de estos pacientes, pudiendo aparecer a cualquier edad, aunque por lo general comienzan en la infancia temprana18. Inicialmente, las crisis son típicamente focales, pero a menudo evolucionan a crisis tónico-clónicas generalizadas. También se pueden encontrar espasmos infantiles, crisis mioclónicas o atónicas. En nuestro estudio la epilepsia apareció en el 100% de los niños, siendo las crisis parciales (simples o complejas) las más frecuentes en el 62% de los casos, seguido de las crisis parciales secundariamente generalizadas (31%) y las crisis generalizadas (8%). El intervalo de edad de la aparición de la primera crisis osciló entre los 2 meses y los 11 años, con una media de 26 meses. El 62% (8 niños) tuvieron la primera crisis con menos de 12 meses, y de estos el 75% sufrieron la primera crisis con 6 meses o menos.
El éxito del control de las crisis con la medicación anticonvulsiva es variable e impredecible. Algunos pacientes tienen largos intervalos sin crisis, incluso sin medicación, mientras que otros tienen ataques frecuentes o prolongados, a pesar de las altas dosis de varios fármacos antiepilépticos. El control de las crisis ha sido muy variable en el grupo de pacientes estudiados, ya que hasta el 31% de los mismos ha necesitado probar más de 3 fármacos para el manejo de la epilepsia, el 15% ha probado solo 3 fármacos, el 31% ha necesitado utilizar 2 fármacos y solo el 23% de los han estado bien controlados con un solo fármaco antiepiléptico.
La aparición de crisis, la edad de inicio de los síntomas y la respuesta al tratamiento afectan el pronóstico. El inicio antes de un año y una mala respuesta a la terapia anticonvulsiva se asocian con una mayor probabilidad de deterioro cognitivo16.
En pacientes con epilepsia asociada con el síndrome de Sturge-Weber se ha sugerido que la epileptogénesis es causada por la isquemia crónica en áreas corticales afectadas por angiomatosis leptomeníngea. Sin embargo, se han encontrado casos de pacientes afectos por este síndrome con epilepsia médicamente intratable en la que el foco epileptógeno era un área de displasia cortical focal tipo IIa cerca de la región de angiomatosis leptomeníngea19,20.
Varios artículos abordan el tratamiento de la epilepsia refractaria en estos pacientes mediante cirugía, en particular realizando hemisferectomías, con mejoría en el control de las crisis21,22. En uno de los pacientes de este estudio se realizó hemisferectomía izquierda (ante la presencia de epilepsia refractaria y síntomas de deterioro cognitivo) con resultados excelentes, ya que hubo una desaparición de las crisis desde el momento de la cirugía hasta la realización de la recogida de datos (7 meses).
La hemiparesia, presente en el 39% de los casos analizados, a menudo se desarrolla de forma aguda en combinación con el inicio de las crisis21. La extremidad parética generalmente no crece a un ritmo normal, lo que produce hemiatrofia en dicho miembro. En algunas ocasiones los niños afectados tienen una pérdida progresiva de la función motora, cuyo mecanismo de producción es desconocido, aunque se postula la existencia de un posible efecto acumulativo de los repetidos episodios trombóticos leptomeníngeos dentro de la malformación, o una alteración crónica del flujo sanguíneo. Numerosos estudios parecen haber demostrado eficacia en la reducción de episodios trombóticos en estos pacientes con el uso de bajas dosis de ácido acetilsalicílico23–25.
Los pacientes con síndrome de Sturge-Weber pueden desarrollar también eventos similares a ictus, llamados episodios stroke-like, en los que se producen déficits neurológicos transitorios que con frecuencia duran entre varios días a semanas18. El 23% de los casos estudiados presentaron uno o varios episodios stroke-like a lo largo de su evolución. Estos episodios pueden ocurrir de forma esporádica, en el contexto de una agrupación de crisis o preceder una migraña o a una crisis. El tratamiento con bajas dosis de ácido acetilsalicílico también ha demostrado eficacia en la disminución de estos episodios23–25. El 54% de los niños de nuestro estudio precisaron tratamiento con antiagregantes por presencia de episodios trombóticos o stroke-like con mejoría subjetiva en la disminución de la frecuencia y gravedad de los mismos.
Las cefaleas, con predominio de migrañas, también aparecen con mayor frecuencia en este síndrome y a edades más tempranas que en la población general18. En nuestra serie de casos se hallaron cefaleas recurrentes en el 39% de los pacientes.
Los niños con síndrome de Sturge-Weber suelen desarrollarse normalmente durante varios meses después del nacimiento, poniéndose posteriormente de manifiesto un retraso en el desarrollo26. La función cognitiva es más pobre en pacientes con lesiones intracerebrales bilaterales27. En nuestra serie encontramos que el 46% de los niños estudiados presentaron signos de retraso psicomotor. Posteriormente estos mismos niños, al realizarles test psicométricos, obtuvieron puntuaciones en el cociente intelectual total por debajo de 70.
El riesgo para los problemas de comportamiento en los pacientes con Síndrome de Sturge-Weber se asocia a una peor función cognitiva y a la presencia de epilepsia. Sin embargo, los problemas de comportamiento también ocurren en pacientes con inteligencia normal28,29.
En los últimos años se han analizado otros posibles síntomas asociados al síndrome de Sturge-Weber, como por ejemplo se ha objetivado que el déficit de hormona del crecimiento aparece hasta 18 veces más en estos pacientes en comparación con la población general. Esto ocurre sin evidenciarse anomalías hipofisarias o hipotalámicas en las pruebas de neuroimagen30. También han sido reportados casos de hipopituitarismo parcial31 e hipotiroidismo central que pueden relacionarse con el uso de determinados anticonvulsivantes32. Puesto que estas condiciones se pueden tratar eficazmente, se recomienda obtener pruebas de detección precoz en estos pacientes.
El diagnóstico de síndrome de Sturge-Weber se basa en la demostración del angioma facial y angiomatosis leptomeníngea. Ocasionalmente, como se ha comentado anteriormente, existen pacientes con malformación intracraneal sin lesión facial que pueden desarrollar también problemas neurológicos.
La técnica de neuroimagen de elección para el diagnóstico es la resonancia magnética con contraste de gadolinio, que es capaz de demostrar la presencia angiomatosis leptomeníngea, y el grado de participación en las estructuras del cerebro33. En algunos casos la participación leptomeníngea no puede ser detectada por técnicas de neuroimagen durante la infancia, y se hace aparente más adelante2. A todos los pacientes de nuestro estudio se les realizó al menos una resonancia magnética a lo largo de su evolución, encontrándose calcificaciones leptomeníngeas en el 85% de los pacientes, todas ellas localizadas en el territorio de la angiomatosis. En controles radiológicos posteriores realizados a estos mismos pacientes se visualizó el aumento de las calcificaciones en el 70% de los mismos.
Si no está disponible la resonancia magnética, la tomografía craneal computarizada puede identificar calcificaciones cerebrales y proporcionar alguna información anatómica. La tomografía computarizada por emisión de fotón único ha demostrado una disminución del flujo en la región cerebral con angiomatosis leptomeníngea18.
Múltiples son los hallazgos radiológicos que pueden ser encontrados en estos pacientes, aparte de los mencionados anteriormente, como son la atrofia de lóbulos o atrofia en otras localizaciones (en nuestra serie de casos la encontramos en 4 pacientes), hipertrofia de plexos coroideos, anomalías en el drenaje venoso, prominencias de las venas colaterales profundas, etc.
El electroencefalograma ofrece un método no invasivo para evaluar la función cerebral que se puede repetir de forma segura y tan a menudo como sea necesario18. La alteración electroencefalográfica más frecuentemente encontrada es la disminución de los voltajes en el hemisferio afecto. Estos hallazgos no concuerdan con los encontrados en nuestro estudio, ya que la alteración más frecuentemente encontrada en los electroencefalogramas de los pacientes estudiados han sido las puntas, puntas-ondas o polipuntas ondas (46%), seguida por zonas de hipovoltaje (31%), puntas ondas asociadas a hipovoltaje (15,5%) y regiones de lentificación (8%).
Estudios en curso con la electroencefalografía están investigando si esta herramienta puede ayudar en el pronóstico y la selección de regímenes de tratamiento18.
Algunos estudios en los niños con síndrome de Sturge-Weber han demostrado que el doppler transcraneal puede ser una herramienta no invasiva útil en la determinación de la gravedad de las anomalías del flujo sanguíneo y la monitorización de los cambios progresivos en el tiempo18.
El desarrollo de biomarcadores vasculares (aumento de metaloproteinasas en orina) se ha informado por Marler et al., que llevaron a cabo la elaboración de perfiles no invasivos de vascularización en pacientes con una variedad de trastornos vasculares, entre los que se encuentran pacientes con síndrome de Sturge-Weber. La continua identificación y validación de biomarcadores para este síndrome será un complemento importante para enfoques terapéuticos futuros18.
No existe un tratamiento específico para el síndrome de Sturge-Weber. Las manifestaciones cutáneas, oculares, neurológicas y endocrinas se tratan con éxito desigual. Es de vital importancia conocer y manejar el tratamiento antiepiléptico en estos niños, puesto que ciertos anticonvulsivantes, como el topiramato, pueden provocar glaucoma agudo bilateral de ángulo cerrado empeorando el pronóstico18.
Existe un consenso cada vez mayor de que dosis bajas de ácido acetilsalicílico pueden ser beneficiosas, con el argumento de que la terapia antitrombótica puede prevenir la progresión de la alteración del flujo sanguíneo cerebral y la lesión hipóxico-isquémica neuronal23–25. Además, hay otros enfoques para la prevención de episodios stroke-like, incluyendo proporcionar una vacunación anual contra la gripe, la hidratación excelente durante una enfermedad, y el tratamiento de la fiebre, pero no han sido rigurosamente estudiados18.
El pronóstico del síndrome de Sturge-Weber depende de la extensión de la angiomatosis leptomeníngea y su efecto sobre la perfusión de la corteza cerebral, así como la gravedad de la afectación ocular. El pronóstico también se ve condicionado por la edad de inicio de las convulsiones y si pueden ser controladas. Las funciones neurológicas pueden deteriorarse con la edad. Como resultado, aproximadamente la mitad de los adultos afectados están deteriorados, incluyendo los que inicialmente fueron normales.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

