



Las alteraciones en el gen PNKP dan lugar a trastornos del neurodesarrollo con grados variables de epilepsia, retraso psicomotor, atrofia cerebelosa y neuropatía periférica1. En la literatura se describen varios cuadros clínicos:
- 1.
Microcefalia, convulsiones y retraso del desarrollo (MIM #613402). Descrita por primera vez por Shen et al. en 2010, de herencia autosómica recesiva. Los pacientes tienen microcefalia congénita, epilepsia precoz con evolución rápida a encefalopatía epiléptica del desarrollo y discapacidad intelectual1–4.
- 2.
Ataxia con apraxia oculomotriz tipo 4 (MIM #616267). Descrito en 2015 por Bras et al. Caracterizado por ataxia y apraxia oculomotora, secundarias a la destrucción cerebelosa. En muchas ocasiones asocian polineuropatía axonal sensitivo-motora. No presentan microcefalia, ni epilepsia3,5,6.
- 3.
En los últimos años se han publicado pacientes con fenotipos intermedios3,4,7–10:
Microcefalia asociada a neurodegeneración y polineuropatía. Fenotipo a medio camino entre los anteriores. Descrito por Poulton et al. en 2013, secundario a la variante homocigota tipo frameshift p.Thr424Glyfs*48, en 2 hermanos con microcefalia congénita, epilepsia, ataxia y degeneración cerebelosa progresiva2,4,7.
Síndrome de Charcot-Marie-Tooth like. Caracterizado por pies cavos, polineuropatía sensitivo-motora y ataxia progresiva. No desarrollan apraxia oculomotora, epilepsia o discapacidad cognitiva8,9.
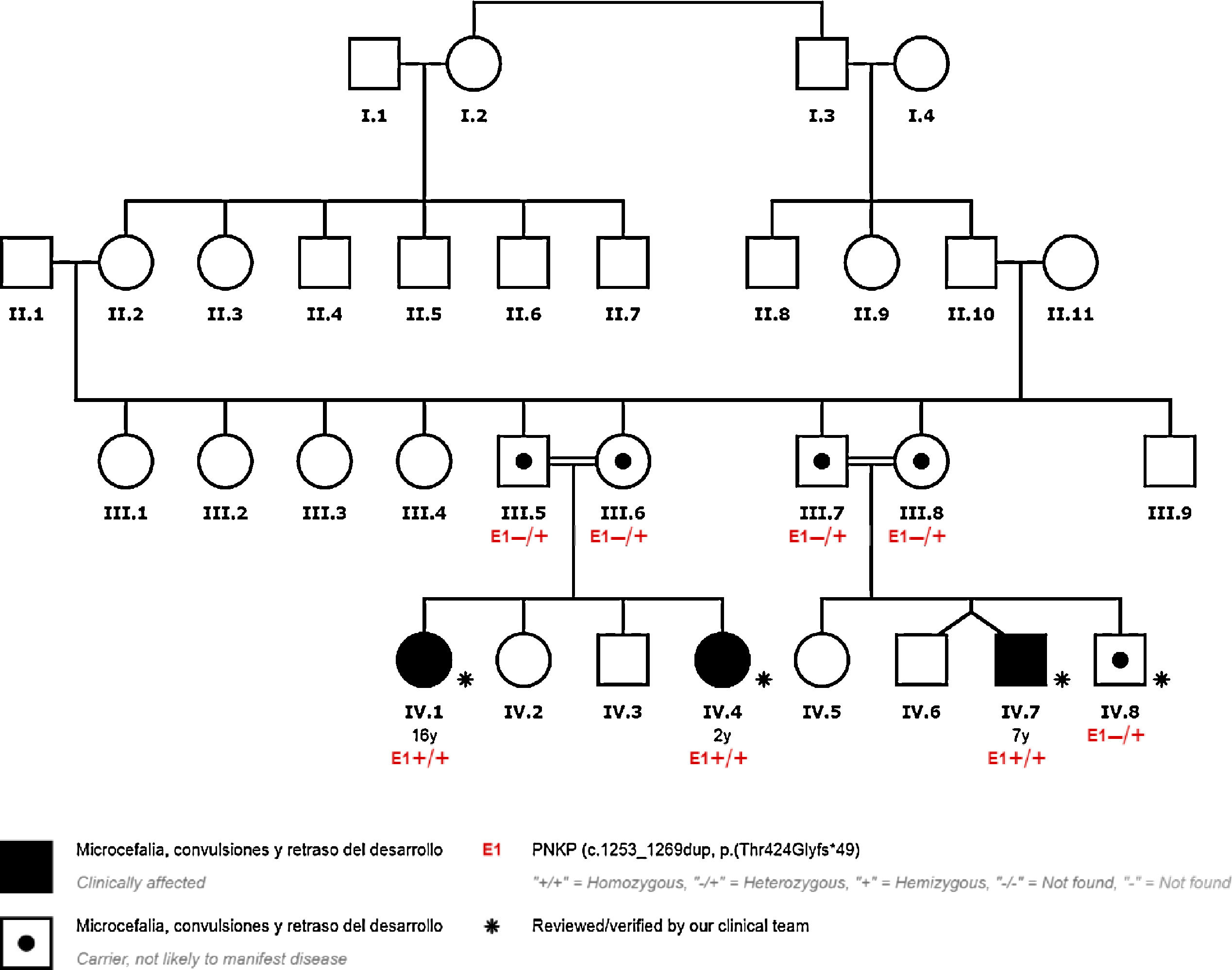
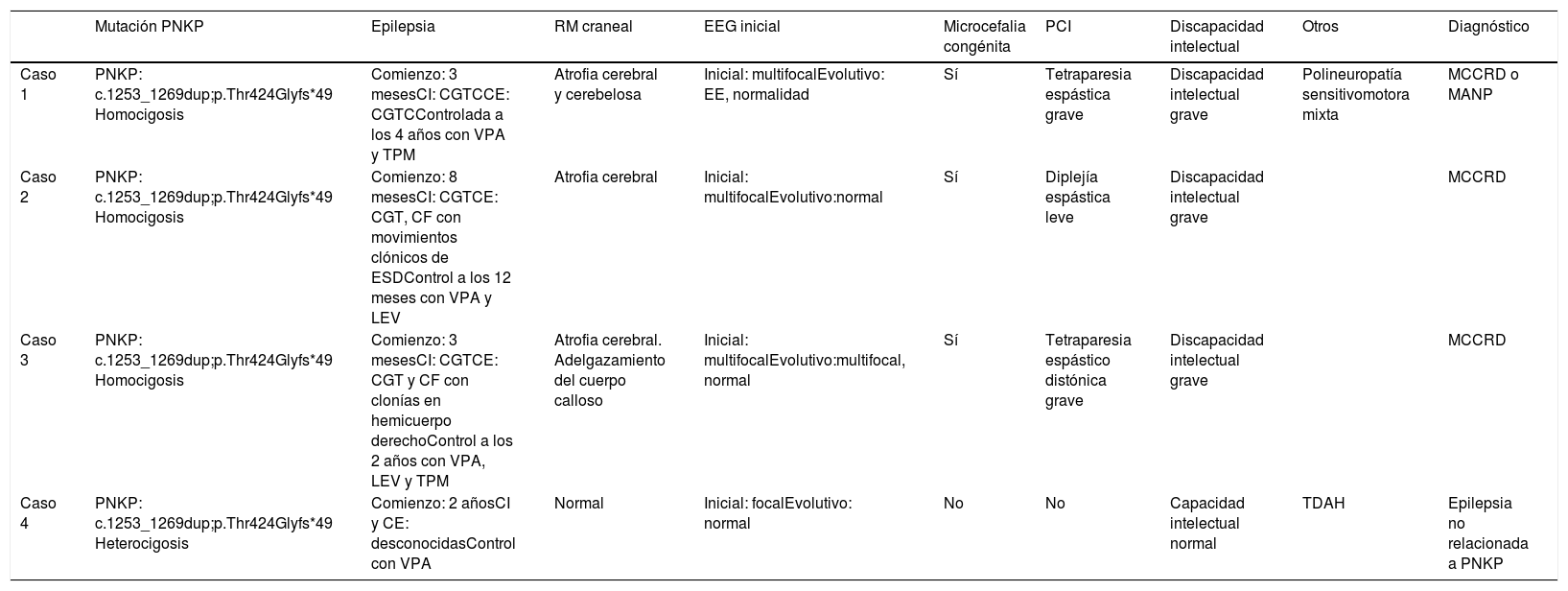
Se presentan los casos de 4 pacientes de una misma familia consanguínea con la variante patogénica en el gen PNKP NM_007254.3:c.1253_1269dup;p.Thr424Glyfs*49, detectada mediante exoma clínico. Se trata de una variante ya descrita previamente en la literatura como patogénica (tabla 1) (fig. 1).
Características de los casos
| Mutación PNKP | Epilepsia | RM craneal | EEG inicial | Microcefalia congénita | PCI | Discapacidad intelectual | Otros | Diagnóstico | |
|---|---|---|---|---|---|---|---|---|---|
| Caso 1 | PNKP: c.1253_1269dup;p.Thr424Glyfs*49 Homocigosis | Comienzo: 3 mesesCI: CGTCCE: CGTCControlada a los 4 años con VPA y TPM | Atrofia cerebral y cerebelosa | Inicial: multifocalEvolutivo: EE, normalidad | Sí | Tetraparesia espástica grave | Discapacidad intelectual grave | Polineuropatía sensitivomotora mixta | MCCRD o MANP |
| Caso 2 | PNKP: c.1253_1269dup;p.Thr424Glyfs*49 Homocigosis | Comienzo: 8 mesesCI: CGTCE: CGT, CF con movimientos clónicos de ESDControl a los 12 meses con VPA y LEV | Atrofia cerebral | Inicial: multifocalEvolutivo:normal | Sí | Diplejía espástica leve | Discapacidad intelectual grave | MCCRD | |
| Caso 3 | PNKP: c.1253_1269dup;p.Thr424Glyfs*49 Homocigosis | Comienzo: 3 mesesCI: CGTCE: CGT y CF con clonías en hemicuerpo derechoControl a los 2 años con VPA, LEV y TPM | Atrofia cerebral. Adelgazamiento del cuerpo calloso | Inicial: multifocalEvolutivo:multifocal, normal | Sí | Tetraparesia espástico distónica grave | Discapacidad intelectual grave | MCCRD | |
| Caso 4 | PNKP: c.1253_1269dup;p.Thr424Glyfs*49 Heterocigosis | Comienzo: 2 añosCI y CE: desconocidasControl con VPA | Normal | Inicial: focalEvolutivo: normal | No | No | Capacidad intelectual normal | TDAH | Epilepsia no relacionada a PNKP |
CE: crisis evolutiva; CGT: crisis generalizada tónica; CI: crisis inicial; CGTC: crisis generalizada tónico-clónica; CF: crisis focal; EE: encefalopatía epiléptica; EEG: electroencefalograma; ESD: extremidad superior derecha; LEV: levetiracetam; MANP: microcefalia asociada a neurodegeneración y polineuropatía; MCCRD: microcefalia con convulsiones y retraso en el desarrollo; PCI: parálisis cerebral infantil; RM: resonancia magnética; TPM: topiramato; VPA: ácido valproico.

Caso 1. Paciente mujer de 16 años. Comienza con epilepsia a los 3 meses y tuvo un estatus convulsivo. La epilepsia se controló a los 4 años. Presentó microcefalia congénita, tetraparesia espástica y discapacidad intelectual. En la resonancia magnética (RM) craneal se visualiza atrofia cerebelosa y engrosamiento difuso del díploe. Clínicamente presenta atrofia de pantorrillas, arreflexia rotuliana y aquílea. En el electromiograma se objetiva neuropatía sensitivo-motora mixta de predominio axonal.
Caso 2. Paciente mujer de 2 años. La epilepsia comenzó a los 8 meses, y presentó un estatus convulsivo al año. Presentó microcefalia congénita, parálisis cerebral infantil y discapacidad intelectual grave. En la RM craneal se visualiza atrofia cerebral difusa.
Caso 3. Paciente varón de 7 años. La epilepsia comenzó a los 3 meses, tuvo varios estatus. La epilepsia se controló a los 2 años. Presentó microcefalia, parálisis cerebral infantil y discapacidad intelectual grave. En la RM craneal se visualiza atrofia cerebral difusa y adelgazamiento del cuerpo calloso.
En los casos 1, 2 y 3 se detectó la variante en el gen PNKP: c.1253_1269dup;p.Thr424Glyfs*49 en homocigosis.
Caso 4. Paciente varón de 8 años. Comenzó a los 2 años con crisis frontales. No presentó microcefalia, ni discapacidad intelectual, ni parálisis cerebral. Neuroimagen normal. Únicamente destacó la presencia concomitante de trastorno por déficit de atención e hiperactividad, con capacidad intelectual normal.
Se detectó la variante en el gen PNKP: c.1253_1269dup;p.Thr424Glyfs*49 en heterocigosis, por lo que se interpreta como portador asintomático. Consideramos que su epilepsia es una fenocopia. Esto quiere decir que comparte características fenotípicas con los pacientes que presentan la mutación en homocigosis, pero estas no pueden atribuirse a su genética.
El estudio de los progenitores de los 4 pacientes demostró que todos eran portadores de dicha variante en heterocigosis.
DiscusiónLos pacientes portadores de la mutación del gen PNKP en homocigosis (casos 1, 2 y 3), presentaron epilepsia precoz, encefalopatía epiléptica del desarrollo, microcefalia congénita, parálisis cerebral infantil y atrofia cerebral. Podrían catalogarse dentro del fenotipo clínico de «Microcefalia, convulsiones y retraso del desarrollo», aunque el caso 1 desarrolló una neuropatía, por lo que cabe plantearse la posibilidad de incluirlo dentro del fenotipo de «Microcefalia asociada a neurodegeneración y polineuropatía».
Con respecto al paciente con la variante del gen en heterocigosis (caso 4), su epilepsia es una fenocopia, que no puede atribuirse a la presencia de la variante en el gen PNKP: c.1253_1269dup;p.Thr424Glyfs*49 en heterocigosis.
Por otra parte, los padres de estos pacientes presentaron la variante del gen en heterocigosis, siendo portadores asintomáticos.
Se han descrito varios cuadros clínicos secundarios a mutaciones en el gen PNKP, algunos con epilepsia y otros sin ella.
A día de hoy se desconoce la razón por la que las mismas mutaciones en el gen PNKP dan lugar a fenotipos diferentes, este fenómeno se conoce como expresividad variable, siendo probable que intervengan otros factores genéticos, epigenéticos y/o ambientales3.
La presencia en los últimos años de fenotipos intermedios conduce a la idea de que todos ellos sean un espectro continuo de la misma enfermedad. Por este motivo ante genotipos complejos cobraría gran importancia la realización de estudios genéticos.
FinanciaciónLa presente investigación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.

