



La variante patogénica recurrente c.625G>A (p.Glu209Lys) en el gen PACS2 ha sido recientemente descrita como causal de encefalopatía epiléptica infantil tipo 66 (OMIM 610423) en 15 pacientes con epilepsia de debut neonatal mayoritariamente, dismorfia facial y disgenesia cerebelosa, asociados o no a otras anomalías del desarrollo, como discapacidad intelectual, rasgos del espectro autista, alteraciones hematológicas y/o anomalías menores distales de miembros1,2. Desde su descripción solo se ha reportado otro caso con fenotipo solapante y variante distinta en ese gen3. Por otro lado, en el año 20124 se describen dos casos de discapacidad intelectual sincrónica por mutación recurrente en el gen PACS1 que comparten características clínicas con los anteriores (hipotonía, epilepsia, dismorfia).
El gen PACS2 se expresa en distintos tejidos y codifica la proteína multifuncional PACS-2, que juega un papel fundamental en la interacción entre la membrana del retículo endoplasmático y la mitocondria. Actúa como un interruptor metabólico que interviene en el tráfico y la señalización entre dichas membranas, así como en la expresión nuclear de genes en respuesta a señales catabólicas o anabólicas5,6. Describimos, por su excepcionalidad, un nuevo caso con la variante recurrente en el gen PACS2 presente en 15/16 pacientes descritos a nivel mundial.
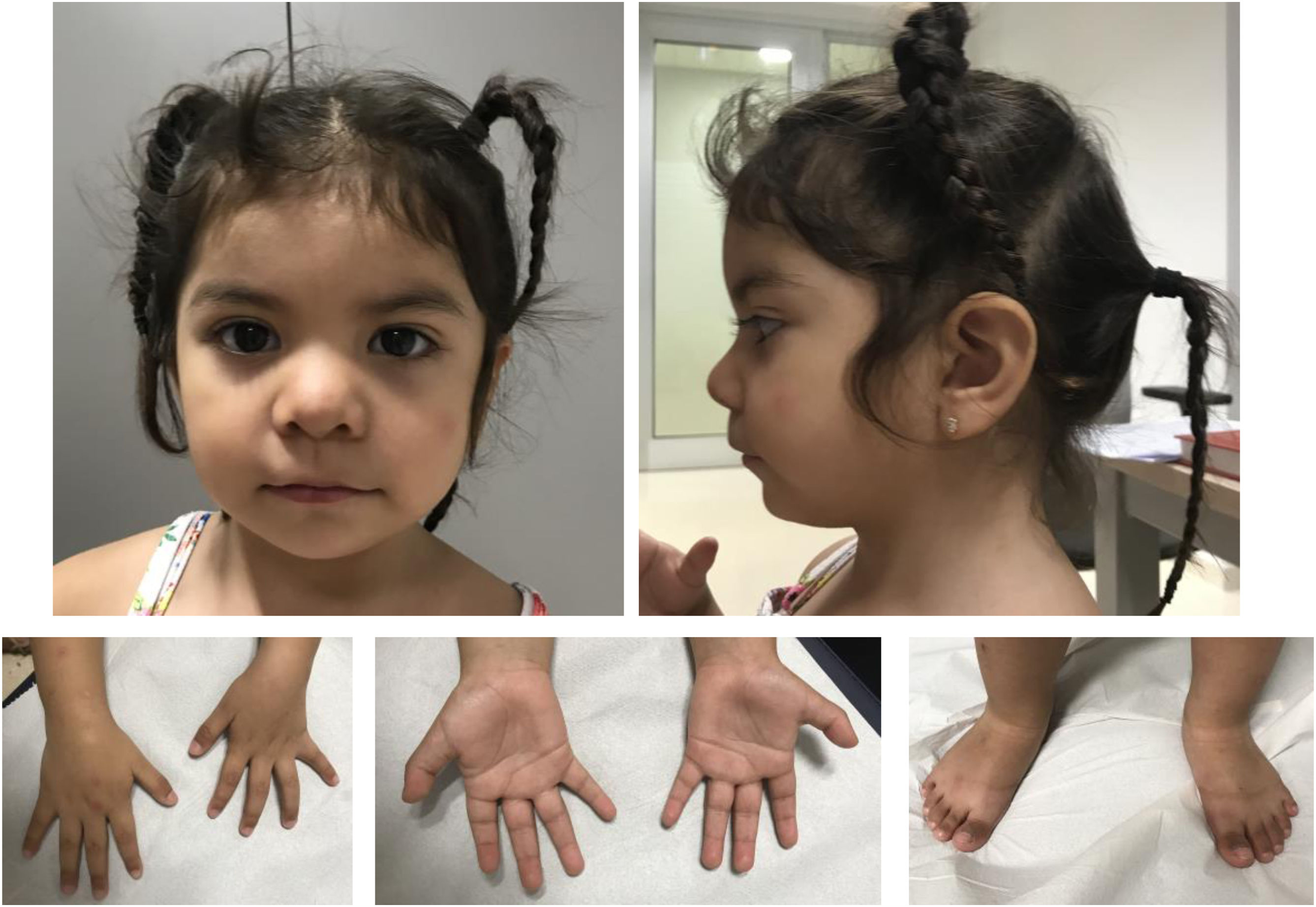
Se trata de una niña de 3años sin antecedentes de interés, segunda hija de padres sanos no consanguíneos de origen boliviano. Fue remitida desde Neuropediatría a consulta de Genética Médica a los 16meses por polidactilia, dismorfia y epilepsia. La gestación fue espontánea y sin incidencias. Parto a término, pesó 4.000g, no precisó reanimación y el periodo neonatal fue normal. El cribado metabólico y las otoemisiones acústicas resultaron normales. Fue intervenida de polidactilia postaxial de ambos pies sin incidencias. Evolutivamente no ha presentado problemas de alimentación y el desarrollo pondoestatural es normal a los 3años: peso: 13,9kg (p63); talla: 89cm (p36); perímetro cefálico: 47cm (p10). La dismorfia facial que presenta es solapante con la descrita previamente, como se aprecia en la figura 1.

Fenotipo clínico a los 3 años de edad. Rasgos toscos, hipertelorismo leve, cejas arqueadas con discreta sinofridia, raíz nasal plana, nariz corta, narinas antevertidas. Labio superior fino con comisuras bucales descendentes. Leve retrognatia. Leve braquidactilia, polidactilia postaxial intervenida en ambos pies.
A los 3meses debutó con crisis consistente en desviación oculocefálica hacia la derecha, hipertonía generalizada y posición distónica de miembros, iniciándose tratamiento con levetiracetam sin respuesta, presentando empeoramiento tras inicio de ácido valproico. Se realizó TC cerebral urgente, que resultó normal, y precisó ingreso en la unidad de cuidados intensivos por estatus epiléptico. Se inició perfusión de midazolam, fenitoína y, tras el control de la crisis, se modificó esta última por zonisamida, que mantiene a día de hoy. El EEG de su ingreso mostró actividad bioeléctrica de base con lentificación difusa, sin apreciarse actividad focal ni otras anomalías epileptiformes. A las 24h los hallazgos electroencefalográficos eran similares pero menos marcados, y al mes el EEG era completamente normal. No obstante, a los 5meses precisó nuevo ingreso por descompensación epiléptica y presentó crisis posteriormente a los 2años y a los 30meses. Permanece sin crisis desde entonces y todos los EEG intercrisis han resultado normales.
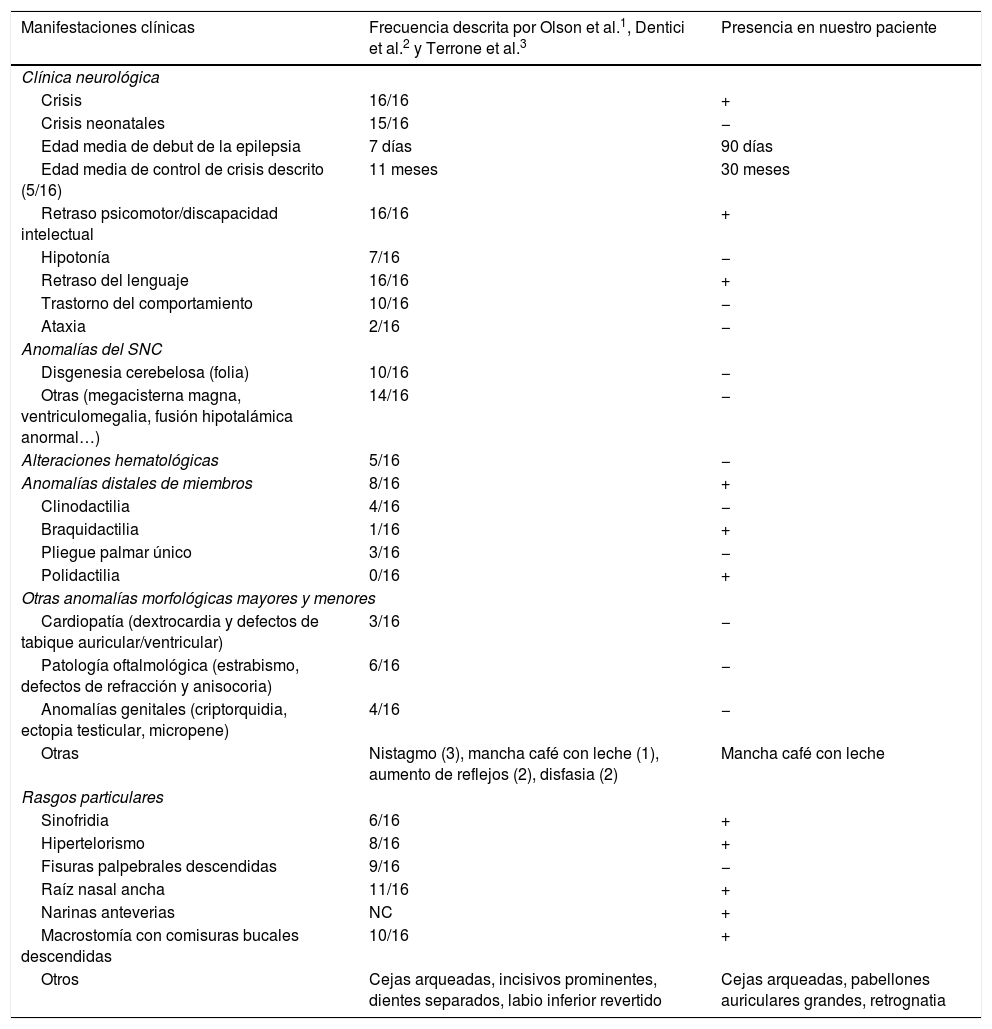
Asocia retraso psicomotor, presentando a los 3años y 6meses de edad, de acuerdo con la escala V.C. Vaughan modificado, una edad de desarrollo de entre 15-18meses en las áreas evaluadas. El lenguaje expresivo es lo más afectado y presenta ocasionalmente conductas auto y heteroagresivas. Está en seguimiento por Atención Temprana y está escolarizara en régimen ordinario con apoyos. En la tabla 1 se muestran los hallazgos clínicos de nuestro caso comparados con los publicados.
Características clínicas de nuestro paciente y los casos publicados
| Manifestaciones clínicas | Frecuencia descrita por Olson et al.1, Dentici et al.2 y Terrone et al.3 | Presencia en nuestro paciente |
|---|---|---|
| Clínica neurológica | ||
| Crisis | 16/16 | + |
| Crisis neonatales | 15/16 | − |
| Edad media de debut de la epilepsia | 7 días | 90 días |
| Edad media de control de crisis descrito (5/16) | 11 meses | 30 meses |
| Retraso psicomotor/discapacidad intelectual | 16/16 | + |
| Hipotonía | 7/16 | − |
| Retraso del lenguaje | 16/16 | + |
| Trastorno del comportamiento | 10/16 | − |
| Ataxia | 2/16 | − |
| Anomalías del SNC | ||
| Disgenesia cerebelosa (folia) | 10/16 | − |
| Otras (megacisterna magna, ventriculomegalia, fusión hipotalámica anormal…) | 14/16 | − |
| Alteraciones hematológicas | 5/16 | − |
| Anomalías distales de miembros | 8/16 | + |
| Clinodactilia | 4/16 | − |
| Braquidactilia | 1/16 | + |
| Pliegue palmar único | 3/16 | − |
| Polidactilia | 0/16 | + |
| Otras anomalías morfológicas mayores y menores | ||
| Cardiopatía (dextrocardia y defectos de tabique auricular/ventricular) | 3/16 | − |
| Patología oftalmológica (estrabismo, defectos de refracción y anisocoria) | 6/16 | − |
| Anomalías genitales (criptorquidia, ectopia testicular, micropene) | 4/16 | − |
| Otras | Nistagmo (3), mancha café con leche (1), aumento de reflejos (2), disfasia (2) | Mancha café con leche |
| Rasgos particulares | ||
| Sinofridia | 6/16 | + |
| Hipertelorismo | 8/16 | + |
| Fisuras palpebrales descendidas | 9/16 | − |
| Raíz nasal ancha | 11/16 | + |
| Narinas anteverias | NC | + |
| Macrostomía con comisuras bucales descendidas | 10/16 | + |
| Otros | Cejas arqueadas, incisivos prominentes, dientes separados, labio inferior revertido | Cejas arqueadas, pabellones auriculares grandes, retrognatia |
Durante su seguimiento por Neuropediatría se realizaron las siguientes pruebas complementarias: RM cerebral y ecografía abdominal (periodo neonatal), sin hallazgos patológicos; valoración cardiológica y oftalmológica normales (no retinopatía); arrayCGH 60k, estudio metabólico en sangre/orina que incluyó sialotransferrinas y esteroles, aminoácidos, ac. orgánicos, lactato/piruvato, perfil de acilcarnitinas, sulfitest, test SAICAR, normal. Tras valoración clínica por Genética, ante la sospecha de trastorno genético específico, posible ciliopatía por la asociación de retraso psicomotor, polidactilia, braquidactilia y dismorfia, se solicitó estudio de genes asociados mediante NGS, sin encontrar variantes candidatas, y evaluación de la audición (potenciales evocados de tronco cerebral y audiometría), que resultó normal. Se amplió estudio a exoma clínico, identificándose la variante patogénica previamente descrita en PACS2. No fue posible el estudio de los progenitores al no estar empadronados en la región.
Se describe un nuevo caso cuya clínica solapa con las formas más leves publicadas al ser el debut de la epilepsia más tardío a lo mayoritariamente descrito y no presentar disgenesia cerebelar. Además se amplía el fenotipo al asociar polidactilia. El empeoramiento clínico tras introducir ácido valproico podría tener relación con la función de la proteína PACS-2, por lo que su uso debería cuestionarse.
Este nuevo caso corrobora la implicación del gen PACS2 en discapacidad intelectual y encefalopatía epiléptica y muestra la gran utilidad del exoma clínico para diagnosticar fenotipos complejos poco frecuentes, recientemente descritos y que están probablemente infradiagnosticados por su desconocimiento.
La variante coincide con la descrita en 15/16 pacientes publicados, lo que corrobora que se trata de una variante recurrente.
El estudio del gen PACS2 debe incluirse en los paneles diseñados para el estudio de las encefalopatías epilépticas, disgenesia cerebelar y ante la sospecha de ciliopatías y/o alteraciones en el gen PACS1.
FinanciaciónNo hay financiación.
Conflicto de interesesNo hay conflicto de intereses en relación con este trabajo.
Damos las gracias a la familia por su colaboración para que esta publicación sea posible.

