



Tubular aggregate myopathy (TAM) and Stormorken syndrome (STRMK) are rare hereditary conditions due to heterozygous mutations in STIM1 (NM_001277961.1) and ORAI1 (NM_032790.3) genes. Both genes play a key role in the store-operated Ca2+ entry (SOCE), regulating calcium homeostasis and thereby control a multitude of Ca2+-dependent pathways including muscle contraction.1 Both entities encompass a broad phenotypic spectrum characterized by progressive muscle weakness and additional features as short stature, facial dysmorphism, platelet dysfunction, asplenia, ichthyosis, dyslexia and miosis, phenotype.2 Meanwhile TAM refers to the muscular disorder associated or not with mild extramuscular features, the full penetrance disorder is referred as STRMK. Most of patients with TAM/STRMK diagnosis carry missense mutations in STIM1 gene while ORAI1 mutations are less frequent.3,4
Herein, we report a case of STRMK, with STIM1 variant c.262A>G (p.S88G) in STIM1 previously described in only one patient,5 in order to support its pathogenic role.
Case reportA 27-year-old woman came to our consultation with a complaint of progressive proximal leg weakness since late adolescence, which she noticed while running and stair climbing.
At the age of 18, proximal weakness started accompanied by knee pain. She was treated then for chondromalacia with pain improvement, but weakness remained stable. One year later, she started to develop bilateral distal weakness with difficulty for feet dorsiflexion, mild scapular weakness and bilateral progressive ptosis.
She had a previous history of nutcracker syndrome treated with left renal vein stenting, cholesteatoma in the left ear, beta thalassemia trait, asplenia and a massive pulmonary thromboembolism secondary to oral contraceptives at the age of 25. She also had a previous history of easy bruising and recurrent epistaxis. Pregnancy, birth history and early development were normal, without hypotonia and with achievement of all motor milestones. She had performed dance and sports until twenties. Cognition was strictly normal. There was no family history of myopathy. Her parents and brother showed no dysmorphic nor myopathic signs and only her mother had beta thalassemia trait.
Clinical examination disclosed short stature, mild scoliosis and scaphocephaly without high arched palate, skin disorders or edema. Neurologic examination showed normal pupillary contraction, mild upward and inward ophtalmoparesis and bilateral ptosis reaching pupillary borders. She had bilateral symmetric facial weakness due to orbicularis oculi and ori muscle affectation, and moderate scapular weakness with winged scapula. Mild proximal arm weakness and moderate to severe proximal and distal leg weakness with limited ankle dorsiflexion were also present. Positive Gowers sign and myopathic gait (Trendelenburg's sign) were noticed.
Blood tests showed increased levels of creatine kinase (608U/L; normal: 24–170/L). Hemogram showed normal hemoglobin level of 13.5g/dl (12.0–16.0) and hematocrit 42.8% (36.0–46.0). Mildly reduced mean corpuscular volume (79.9fL; normal: 82.0–97.0fL) and mean corpuscular hemoglobin (25.9pg; normal: 26.0–31.0pg), as well as increased red blood cell distribution width (15.2%; normal: 8.0–14.8%), with normal reticulocytes count (0.06×106/μL; normal 0.03–0.09×106/μL). These findings were probably due to beta thalassemia trait however peripheral blood smear was not performed. The remainder results, including serum calcium (8.5mg/dL), platelet count (284.0×109) and standard coagulation study (PT, TA, TTPA and INR) were normal.
Nerve conduction studies were strictly normal. Needle electromyography disclosed diffuse myopathic changes which were more expressive in both maximum and medium gluteus and both iliopsoas muscles. Due to these findings a deltoid muscle biopsy was performed.
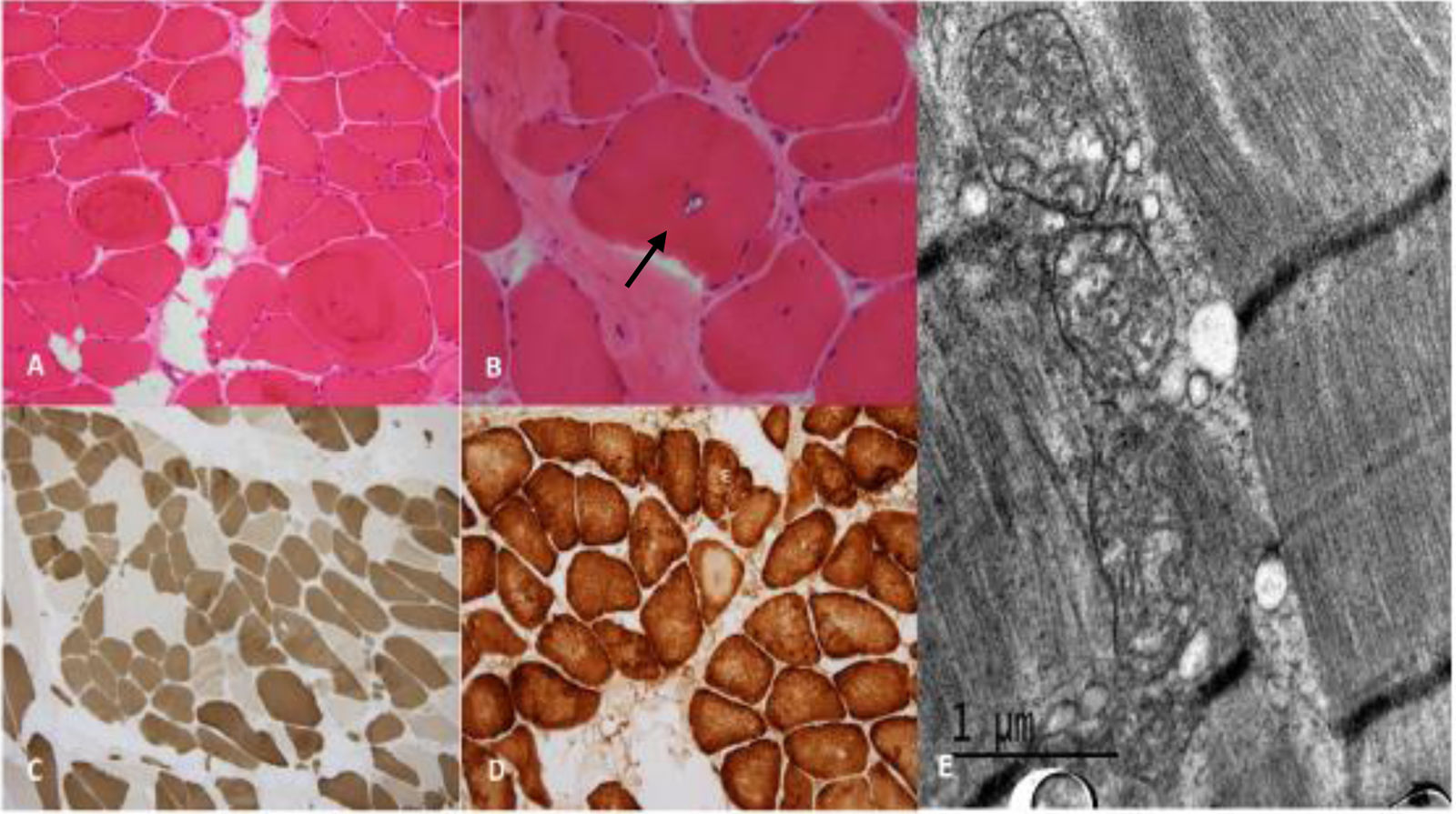
Histologic study revealed marked variation of muscle fibers size and shape with nuclear internalization. Mild increase in endomysial connective tissue was present. Rimmed vacuoles were identified in some muscle fibers. Scared necrotic fibers were observed though there was no inflammatory cell infiltration. Tubular aggregates (TA) or sarcoplasmic inclusions were not observed. ATPase staining showed predominance of type 1 fibres. Oxidative techniques revealed absence of staining in central core-like areas in some muscle fibers. There were no cytochrome c oxidase (COX) negatives fibers. No abnormalities were observed with Periodic Acid-Schiff (PAS) or oil red staining. The major histologic and histochemical findings are shown in Fig. 1.
 shows increased variability in size and shape of muscle fibers and nuclear internalization (A). Rimmed vacuoles (arrow) are identified in some muscle fibers (B). In myosin ATPase-reacted section (200×) staining shows a mosaic pattern though with a predominance of type 1 muscle fibers (C). COX staining (200×) shows absence of staining in central core-like areas in some fibers (D). Electron microscopy shows mitochondria with elongated cristae but tubular aggregates were not found (E).")
Histologic and histochemical study. Light microscopy, frozen sections. The H&E stain (A 200×, B 400×) shows increased variability in size and shape of muscle fibers and nuclear internalization (A). Rimmed vacuoles (arrow) are identified in some muscle fibers (B). In myosin ATPase-reacted section (200×) staining shows a mosaic pattern though with a predominance of type 1 muscle fibers (C). COX staining (200×) shows absence of staining in central core-like areas in some fibers (D). Electron microscopy shows mitochondria with elongated cristae but tubular aggregates were not found (E).
The electron microscopy analysis revealed an increase in the number of mitochondria with elongated and dilated cristae, without presence of paracrystalline inclusions. No tubular aggregates or other pathological changes were observed. The mitochondrial genetic study (point mutations, rearrangements and deletions by Southern blot) and the respiratory chain were normal.
Transthoracic echocardiography, electrocardiogram and pulmonary tests were also performed without any abnormal features.
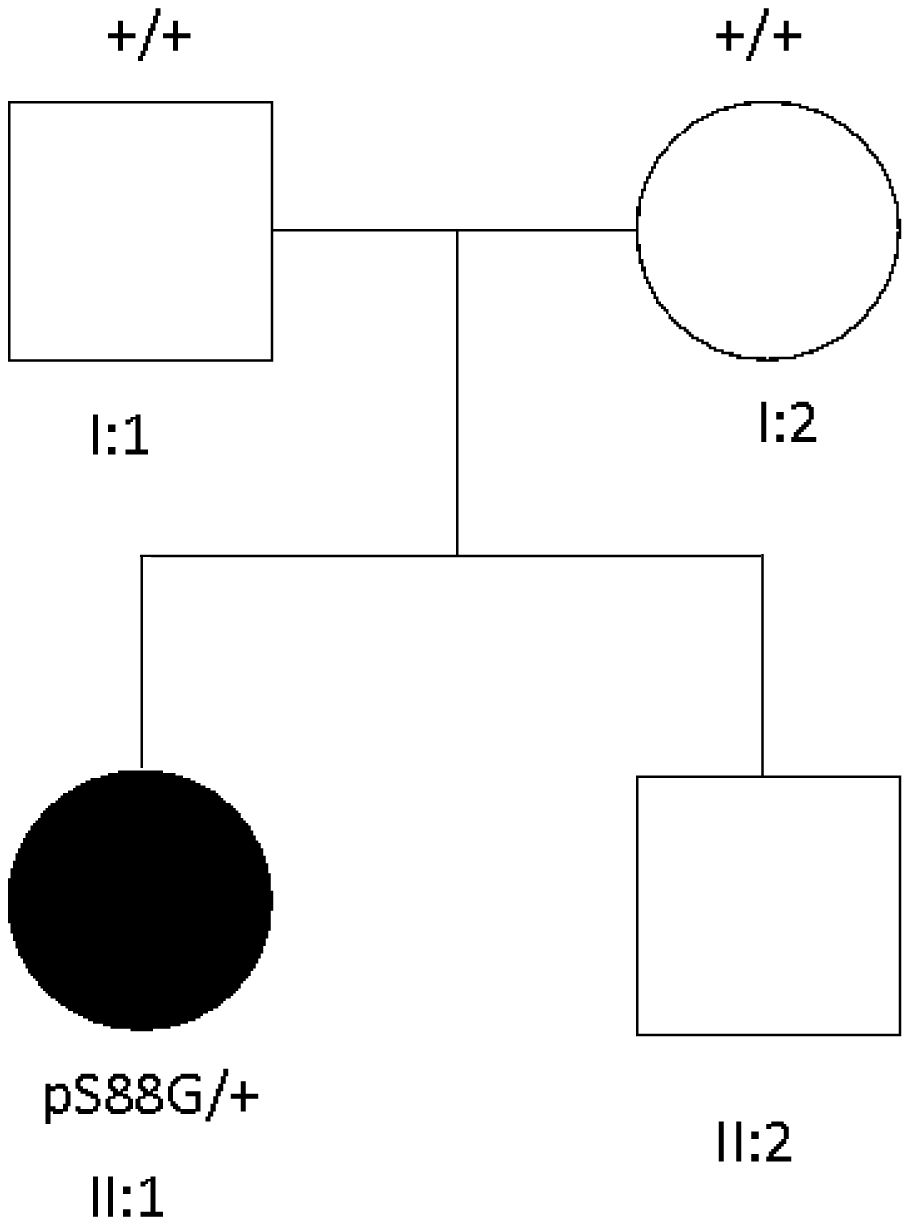
Trio-based exome sequencing was performed in the patient and her parents. A de novo heterozygous missense variant c.262A>G was detected in exon 2 of the STIM1 gene, consistent with the diagnosis of STRMK. Pedigree of the family is shown in Fig. 2.
, which causes a pS88G aminoacid substitution. No other family members are affected.")
At 2 years follow-up visit, the patient remained stable with no weakness progression and genetic counselling was provided for her family.
DiscussionSTIM1 is the main Ca2+ sensor of the endoplasmic reticulum (ER). The protein contains a luminal N-terminal region, a single transmembrane domain and a cytosolic C-terminal region. The N-terminal is formed by two highly conserved EF-hand domains (EF1 and EF2) acting as calcium sensors and a sterile α-motif domain (SAM), whereas the C-terminal domain contains three coiled-coil (CC1-3) domains. In absence of calcium, STIM1 undergoes oligomerization, and binds the sarcoplasmic Ca2+-release-activated Ca2+ channel (CRAC), ORAI1 promoting calcium influx into the cell.6 Missense variants in EF-hand domains act as gain of function (GOF) mutations, preventing ER calcium binding and promoting STIM1 oligomerization and cytosolic calcium entry.7
EF-hand domain mutations are associated with a broad clinical spectrum which ranges from asymptomatic carriers to normally childhood-onset muscle weakness with or without other features such as eye movement defects or contractures and one or more signs of STRMK.2 Most of these mutations are associated with a childhood-onset muscle weakness, although adolescence onset (two patients with p.D84G and p.H109N mutations), and adult onset (six patients) have previously been reported.2,8
To our knowledge, the variant c.262A>G (p.S88G) in the EF-hand domain has only previously been described in one single patient, with a de novo heterozygous mutation and clinical features of progressive upper and lower limbs weakness started in early childhood, contractural phenotype with rigid spine, generalized muscle wasting, and other conditions such as high arched patalate, dry skin, easy bruising, anisopoikilocytosis consistent with hyposplenism and small spleen in ultrasound. Patient also present cardiac involvement with second-degree conduction block. Muscle biopsy of this patients showed marked dystrophic features with fiber size variation and multiple hypertrophic fibers with rimmed vacuoles. Like our patient, there were not tubular aggregates except for intense NADH staining in a several fibers.5
By contrast our patient presented with a milder phenotype including adolescence onset upper and lower limbs weakness, winged scapula and mild ophtalmoparesis. No contractures were observed and myalgia was not a relevant symptom. Asplenia was the feature more consistent with STRMK phenotype. No ichthyosis or platelet count alteration was found in our patient, although a subjacent thrombotic disorder associated to the genetic condition may be responsible of the massive pulmonary embolism previously presented with precipitation in the context of oral contraceptive and the past history of easy bruising and recurrent epistaxis. Anisopoikilocytosis was not investigated in our patient. Short stature, mild scoliosis and scaphocephaly was also noted, being the first time in literature that this craniosynostosis is described in STRMK. Although scaphocephaly is not an unusual condition, it is known that elements of the SOCE (Orai1 and Stim1 proteins) play a crucial role in the differentiation of osteoclasts and osteoblastic function, thus being able to give rise to an excess of bone remodeling activity during embryogenesis that may therefore produce bone alterations in the form of short stature and craniosynostosis.9,10
The phenotypic differences between our patient and the previously reported show the interfamilial variability present in STIM1 EF-hand mutations. This phenomenon has already been studied and for example the EF-hand mutation c.216C>A (p.H72Q) has been described in four different families with different phenotypes ranging from asymptomatic hiperCKemia to complex STRMK phenotypes, indicating possible implication of modifier genes and/or non-genetic factors.2,8 In addition, as in the previously reported patient, we did not observe TA in the biopsy. Even TA are major hallmark of TAM/STRMK spectrum, the Stim1R304W+/− murine model of Rojas-Silva, did not show this finding in the muscle biopsy either while on the contrary it showed same changes in fiber size variability, internalized nuclei and type I fiber predominance as our patient.11 This indicates that the formation (or the timepoint of formation) of TAs may be mutation-dependent and that the presence/absence of TAs does probably not correlated with disease severity.
STIM1 variant c.262A>G has been recognized as a variant of uncertain significance (VUS) in ClinVar database based on a single consistent STRMK phenotype, absence on The Exome Aggregation Consortium (ExAC) and genomAD database, and predicted pathogenic effect by in silico studies. As this patient report is also consistent with a STRMK phenotype, we consider that c.262A>G variant should be updated as pathogenic or probably pathogenic variant in STIM1 gene.

