



Alemtuzumab is a highly effective drug approved by the European Medicines Agency as a disease-modifying drug for the treatment of relapsing-remitting multiple sclerosis.
ObjectiveA consensus document was drafted on the management of alemtuzumab in routine clinical practice in Spain.
DevelopmentA group of multiple sclerosis specialists reviewed articles addressing treatment with alemtuzumab in patients with multiple sclerosis and published before December 2017. The included studies assessed the drug's efficacy, effectiveness, and safety; screening for infections and vaccination; and administration and monitoring aspects. The initial proposed recommendations were developed by a coordinating group and based on the available evidence and their clinical experience. The consensus process was carried out in 2 stages, with the initial threshold percentage for group agreement established at 80%. The final document with all the recommendations agreed by the working group was submitted for external review and the comments received were considered by the coordinating group.
ConclusionThe present document is intended to be used as a tool for optimising the management of alemtuzumab in routine clinical practice.
Alemtuzumab es un fármaco de alta eficacia aprobado por la Agencia Europea de Medicamentos como tratamiento modificador de la enfermedad en pacientes con esclerosis múltiple remitente recurrente.
ObjetivoElaborar un documento de consenso sobre el manejo de alemtuzumab en la práctica clínica habitual, que sea de aplicación en el ámbito español.
DesarrolloUn grupo de expertos en esclerosis múltiple revisó las publicaciones disponibles hasta diciembre de 2017, de tratamiento con alemtuzumab y esclerosis múltiple. Se incluyeron trabajos sobre eficacia, efectividad y seguridad, despistaje de infecciones y vacunación, administración y monitorización. La propuesta inicial de recomendaciones fue desarrollada por un grupo coordinador con base en la evidencia disponible y en su experiencia clínica. El proceso de consenso se llevó a cabo en 2 etapas; se estableció como porcentaje inicial de acuerdo grupal el 80%. El documento final con todas las recomendaciones acordadas por el grupo de trabajo se sometió a revisión externa y los comentarios recibidos fueron considerados por el grupo coordinador.
ConclusionesEl documento aportado pretende ser una herramienta útil para facilitar el manejo del fármaco en condiciones de práctica clínica habitual.
The growing number of therapeutic options for multiple sclerosis (MS) and their increasing efficacy have led to a rise in the complexity of treatment management. Consequently, there is a need for consensus documents that standardise the use of these treatments and enable adequate interpretation of their effectiveness, monitoring, and benefit/risk balance.
Alemtuzumab is a recombinant humanised monoclonal antibody which was approved by the European Medicines Agency (EMA) in September 2013 as a disease-modifying treatment for relapsing-remitting multiple sclerosis (RRMS); it was first marketed in March 2015.1
The high efficacy of alemtuzumab in treating MS, its innovative action mechanism, and the increasing level of management required in its administration have given rise to the need for an in-depth analysis regarding its use. To date, several authors have published recommendations and guidelines on specific aspects of the management of alemtuzumab.2–5
The main aim of this consensus statement is to contribute to the knowledge of the practical management of alemtuzumab, addressing questions related to effectiveness, tolerability, and the management of possible adverse effects, as well as the selection of candidate patients. This consensus document includes recommendations based on evidence from observational studies, case series, and expert recommendations.
Action mechanismAlemtuzumab is a humanised monoclonal antibody that recognises CD52, a molecule that is abundantly expressed in mature T and B lymphocytes but not in their precursors, and that is expressed in very low amounts in innate response cells such as granulocytes.6,7
Alemtuzumab binds to CD52 molecules present in the membrane of circulating lymphocytes in the peripheral blood and induces their lysis via complement action or via antibody-mediated cytotoxicity. This induces marked lymphocytopaenia; the drug's effect is less pronounced in secondary lymphoid organs such as the lymph nodes and the spleen, and nearly absent in bone marrow.8 The preservation of lymphoid progenitors in bone marrow enables the reconstitution of blood lymphocyte populations. During this reconstitution, the new immune response is reprogrammed, with an increase in the ratio of regulatory to effector cells, a marked increase in the production of anti-inflammatory cytokines, and a decrease in proinflammatory cytokines.9,10 This induces prolonged stabilisation or improvement of the disease in a high percentage of patients, despite occasionally provoking new autoimmune pathology. The preservation of the innate immune response, and the fact that lymphocytes not eliminated by the treatment remain fully functional,11 may account for the low rate of severe infections after treatment with alemtuzumab.
In summary, alemtuzumab causes depletion of circulating lymphocytes, with a subsequent repopulation that instigates a change in the pathological inflammatory response towards a more tolerogenic phenotype. As a result, a high percentage of patients present prolonged disease remission.
Clinical development of alemtuzumabThe efficacy and safety of alemtuzumab 12mg have previously been evaluated in 3 clinical trials in which it was compared against subcutaneous interferon beta-1a (44mg) in patients with active RRMS. The first 2 trials included treatment naïve patients (CAMMS223, phase II,12 3-year duration, and CARE-MS I, phase III,13 2-year duration), while the third was based on patients showing inadequate response to previous treatment (CARE-MS II, phase III,14 2-year duration).
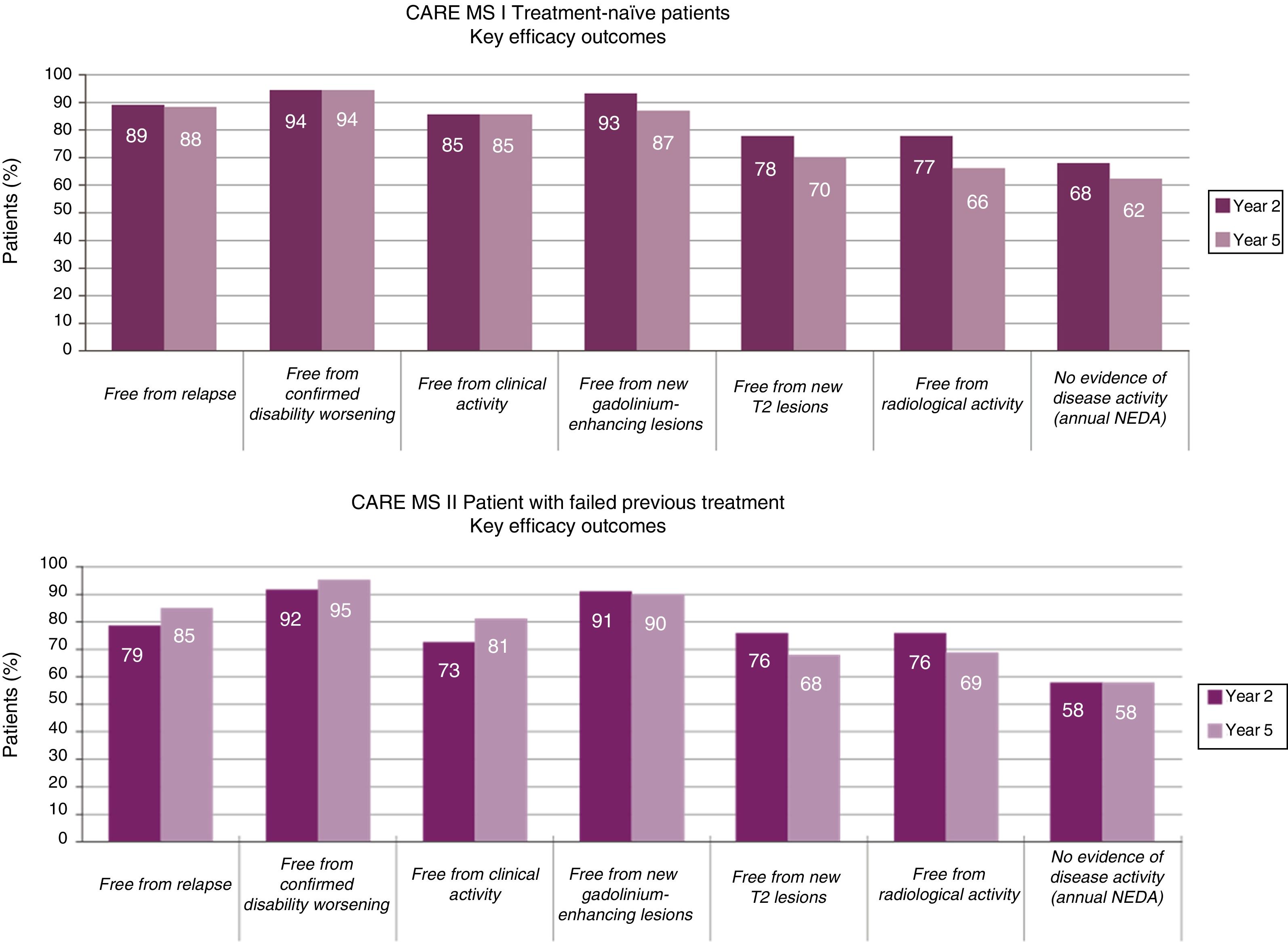
Effectiveness of alemtuzumabEffectiveness in randomised trialsAccording to data from these studies, patients treated with alemtuzumab 12mg showed a significant reduction in relapses (69%,12 55%,13 and 49% of patients,14 respectively; P<.001), as well as a significant reduction in confirmed disability progression (hazard ratios 0.25 [P<.001]12 and 0.58 [P=.008]),12,14 compared to patients receiving interferon beta-1a (the CARE-MS I study showed a hazard ratio of 0.70 [P=.22]).13Table A.1 (Appendix A) shows the main clinical and magnetic resonance imaging (MRI) results of the two CARE-MS pivotal studies.13,14
Long-term effectiveness in extension studiesIn the CARE-MS extension studies (5 years of follow-up), the patient retention rates were 96%15 and 91%16 from the beginning of the extension phase, and 89%15 and 82%16 from the beginning of the pivotal studies.17 Given these high retention rates, data on the efficacy of alemtuzumab are very robust when compared to long-term studies into other disease-modifying treatments.18–20
The reduction observed in the annual relapse rate persisted over time, shifting from 0.18 and 0.28 at the end of the pivotal studies (year 2, cumulative annual rate for years 0-2)13,14 to 0.16 and 0.21 in the extension studies (year 5, cumulative annual rate for years 3-5)15,16 (Fig. 1).

At 5 years, 94%-95% of patients remained free of confirmed disability progression and 33%-43% showed confirmed disability improvement (CDI), over a period of 6 months.15,16 The data demonstrate an increase in the proportion of patients with CDI from year 2 to year 5 (CDI in years 2, 3, 4 and 5: 24%, 28%, 30%, and 33% in CARE-MS I and 29%, 35%, 41%, and 43% in CARE-MS II).15,16
Data on MRI lesions (in gadolinium-enhanced T1-weighted and T2-weighted sequences) from the extension studies showed an absence of new lesions or worsening of existing ones in >75% of patients in all studies (Fig. 1).15,16
The annualised percentage of patients displaying “no evidence of disease activity” (NEDA) status (composed variable of: absence of clinical [relapses or disease progression] or radiological progression [absence of lesions with contrast uptake, new lesions, or lesions increasing in size on T2 sequences]) remained stable at approximately 60% throughout the extension studies.15,16
The results of the TOPAZ study (joint extension of CARE-MS I and II after 7 years of follow-up), presented at the 7th Joint ECTRIMS-ACTRIMS Meeting, consolidates the previous findings on efficacy in the absence of continued treatment, both in treatment naïve patients and non-responders to previous disease-modifying treatments, showing a sustained reduction in MRI activity and loss of brain volume21,22 and an improvement in clinical variables.23,24
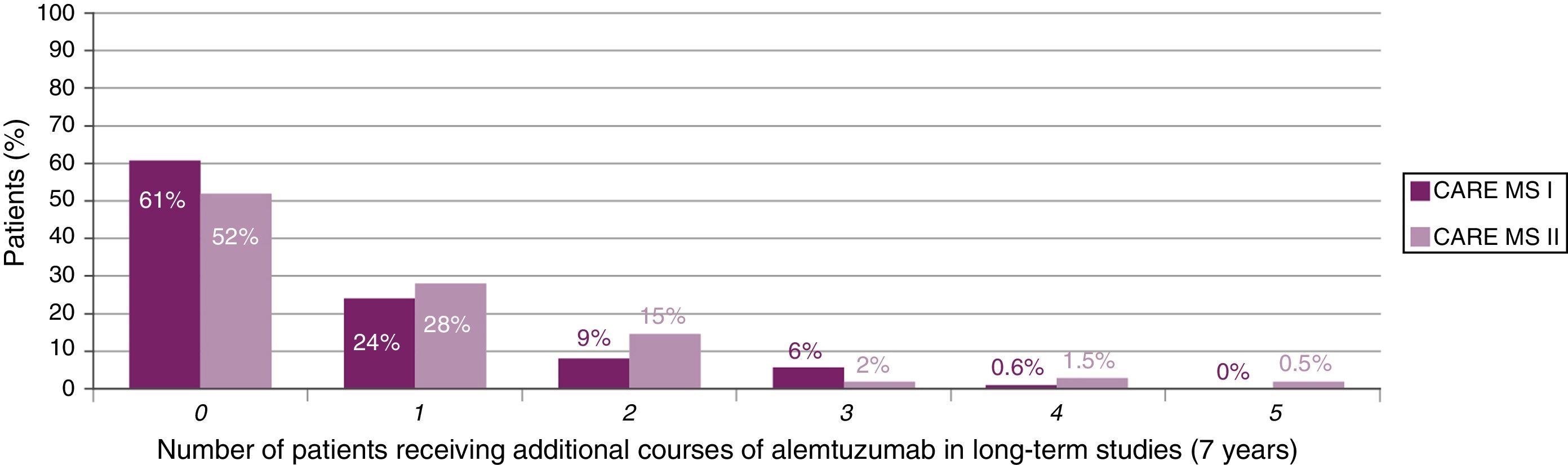
The majority of patients did not receive additional courses of alemtuzumab after the second course (Fig. 2). In those who did require additional courses, the reasons, over a 7-year period, were (for CARE-MS I and II, respectively) relapse in 50% and 57% of patients, radiological activity in 28% and 19%, and both in 20% and 21%.23,24

Clear long-term benefits are reported for treatment with 2 courses of alemtuzumab in patients presenting relapses between the first and second courses.25,26
New data suggest that a third course of alemtuzumab also has clear benefits for patients presenting disease activity after the first 2 courses.25,27
Safety data for alemtuzumabSafety in randomised trialsAlemtuzumab has a good safety profile, with an effective protocol for detecting and treating adverse events.
The CAMMS223, CARE-MS I and CARE-MS II studies mainly report reactions associated with perfusion (> 90%), infections (66%, 67%, and 77%, respectively), and secondary autoimmune diseases, with thyroid disorders in 26%, 16%, and 18% of patients, immune thrombocytopaenia in 1.9%, 1%, and 1%, and nephropathies in 0.3% of patients (all 3 groups).12,14
Long-term safety in extension studiesAlemtuzumab presents good long-term safety, with the annual incidence of adverse events decreasing after the first year of treatment.
The exposure-adjusted incidence rate for all adverse events was lower in the extension studies than in the pivotal studies.15,16
Most adverse events were of mild or moderate severity; no study participant dropped out due to adverse events.15,16 Adverse events related to perfusion were minor and none were serious. The main reactions associated with perfusion were headache, pyrexia, and rash.15,16 The incidence of infections also did not increase, and most were mild or moderate (99.2% and 97.7%).15,16 The most frequent reactions were nasopharyngitis, urinary tract infection, upper respiratory tract infection, and herpes infections.15,16
The most common autoimmune adverse events were thyroid disorders, with peak incidence occurring in the third year (16.7% and 16.5%).15,16
The 7-year safety studies point to a reduction in adverse events compared to pivotal studies. The incidence of thyroid disorders and infections was 1.6% and 30.8% in year 7, vs. 6.4% and 56.1% in year 1.23 Similar results are reported for patients not responding to previous disease-modifying treatments (CARE-MS II).24
Safety findings from the CAMMS223 extension study (10-year follow-up) are consistent with those from the main study.28
Consensus processA formal consensus methodology was followed in the preparation of this consensus statement (modified nominal group technique).29 The consensus process was performed in two stages and a threshold of 80% agreement was established a priori for accepting a recommendation.
During the first stage, participants were sent an email containing a summary of the available evidence on the different priorities for the statement (efficacy, safety and effectiveness, infection screening, vaccination, administration, and monitoring) and a list of possible recommendations proposed by the multidisciplinary panel, based on the evidence available and their clinical experience. Participants also received a general explanation of the methodology and a form for classifying their level of agreement on a 9-point Likert-type scale: 1-3 (inappropriate strategy), 4-6 (uncertain), and 7-9 (appropriate strategy) (Appendix B). During this phase, recommendations were submitted to a first round of individual voting in which all members of the working group used this scale to rate their level of agreement. Participants were also able to add new recommendations or modify the proposed ones in the event of disagreement.
Subsequently, the working group received an email including a summary of the available evidence about the proposed recommendations and the percentage of agreement during the first round (Appendix B).
During the second stage, the members of the coordinating group and the working group met at a face-to-face consensus meeting led by 2 moderators belonging to the coordinating group (XM and OF). At the meeting, the results of the first round of voting were presented anonymously. Recommendations with less than 80% agreement were redrafted, and a second round of voting was carried out by show of hands. Those recommendations that could not be approved during the face-to-face meeting due to time constraints were subsequently agreed upon via email. Finally, recommendations not approved during this stage were excluded from the consensus statement.
RecommendationsSelection of candidate patients for treatment with alemtuzumabAccording to the summary of product characteristics, alemtuzumab can be used in adult patients with RRMS presenting clinical or radiological disease activity.30 In the CARE-MS II study, the most frequent disease-modifying treatments previously used were interferon beta and glatiramer acetate; natalizumab only accounted for 3% of previous treatments and fingolimod had not been approved at the time of the trial.14 Therefore, the current evidence on the management of patients receiving alemtuzumab after treatment with fingolimod, natalizumab, and other immunosuppressants is mainly from published case series from clinical practice.31
Recommendation 1If we combine the recommendations of the summary of product characteristics and the baseline clinical profiles of patients included in the CARE-MS I and II clinical trials, we may conclude that the characteristics of the candidate patient for alemtuzumab should be similar to the following: clinical or radiological disease activity and/or suboptimal response to a full course of disease-modifying treatment and/or rapid and aggressive disease progression.
Recommendation 2Although the use of alemtuzumab in patients with MS previously receiving selective disease-modifying treatment with immunosuppressants has not been associated with safety problems in clinical case series published to date, additional safety measures should be adopted beyond those established in the summary of product characteristics in patients who have previously been treated with other immunosuppressants.
Questions to be considered before treatment onsetInfections associated with the use of alemtuzumabThere was no increase in the incidence of severe or opportunistic infections in the patients included in the CARE-MS I and II studies. However, cases were described of patients with active tuberculosis (0.3%), mainly from endemic areas. Clinical series report isolated cases of listeriosis,32,33 nocardiosis of the central nervous system,34 pneumonitis,30 and pneumonitis associated with pericarditis.35 No cases of progressive multifocal leukoencephalopathy (PML) have been reported. No data are available on the potential reactivation of chronic infections due to hepatitis B or C virus as these patients were excluded from the clinical trials.
No specific risk factors for infection have been identified in patients with RRMS treated with alemtuzumab. Although there is a somewhat higher incidence rate in the first month after administration of the drug, infections are distributed evenly over the follow-up period and tend to decrease over time.36 Lymphocyte count (total and CD4+ T lymphocytes) has not been correlated with the appearance of infections.37
Infections preventable by vaccinationVaccination of patients with MS receiving treatment with immunosuppressants or immunomodulators is part of the multidisciplinary management of the disease. The effect of these drugs on the immune system may increase the risk of such infections as encephalitis and meningitis due to herpes simplex virus or varicella zoster.38,39
From a legal perspective, indicating vaccination in these patients in accordance with official vaccination recommendations does not require written informed consent.
Recommendation 3Vaccination early in the course of the disease is recommended to ensure greater vaccine response. For instance, it has been shown that in the case of vaccination against pneumococcus, immunogenicity is lower in patients receiving immunosuppressants in monotherapy.40 As a rule, live attenuated vaccines are contraindicated in pregnant women and in patients receiving immunosuppressants. On the contrary, there is no contraindication for inactivated vaccines in patients receiving disease-modifying treatment,41 although it is important to assess the drug's mechanism of action to select the optimal moment for vaccination, with a view to ensuring the greatest protective effect.
Vaccination strategies with alemtuzumabRecommendation 4It is advisable to evaluate the need for vaccination before starting treatment with alemtuzumab. To this end, it is essential that we (1) know the patient's serological status against measles, varicella, hepatitis A, and hepatitis B; and (2) document the patient's vaccination history, with special emphasis on seasonal influenza, pneumococcus, hepatitis A and B virus, tetanus/diphtheria, varicella, measles and human papillomavirus.
Recommendation 5It is advisable to evaluate vaccination in patients starting alemtuzumab treatment, as specified in Table 1. The table describes the vaccines to be evaluated, their composition, and the pattern of both primary vaccination and booster doses.
Vaccinations to consider before alemtuzumab administration.47,48
| Vaccine | Type | Indication | Schedule | Booster dose |
|---|---|---|---|---|
| Seasonal influenza | InactivatedFractional or subunits | Yes | 1 annual dose during influenza vaccination campaign (October-December) | Not required |
| Pneumococcal 13-valent | InactivatedProtein-conjugate polysaccharide | Yes | 1 dose | Not required |
| Pneumococcal 23-valent | InactivatedPolysaccharide | Yes | 1 doseAt least 2 months after administration of the pneumococcal 13-valent conjugate vaccine | At 5 years |
| Hepatitis Ba | InactivatedSurface antigen | Yes, provided anti-HBs<10IU/mL | 0-1-2-6 monthsd | Not required |
| Hepatitis A | InactivatedWhole viruses | Yes, provided IgG HAV (–) | 0-6 months | Not required |
| Tetanus/diphtheria | InactivatedTetanus toxoid | Yes. Primary vaccination should be started if not received in the past, or complemented with booster dosing | Primary vaccination (3 doses):0-1-6 months or 0-1-12 months | Two doses: the first should be administered 10 years after primary vaccination and the second 10 years after the first booster dose |
| Human papillomavirus | InactivatedSubunits | Yesb | 0-1-6 months or 0-2-6 months | Not required |
| Varicellac | Live attenuatedWhole virus | Yes, provided IgG varicella (–) | 0-1 month | Not required |
| Triple viral (Measles, mumps, and rubellac) | Live attenuatedWhole virus | Yes, provided IgG measles (–) | 0-1 months | Not required |
Anti-HBs: Hepatitis B surface antibody; HAV: Hepatitis A virus; Ig: immunoglobulin.
Clinicians may consider administering AS04C adjuvanted vaccine, high-load (40μg) HBV vaccine, or double dose of the conventional vaccine, maintaining the same vaccination schedule.
Preferably in women under 26 years of age who have not received the vaccine according to the systematic calendar for children. Also in women scheduled to undergo cervical excision procedures due to high-grade lesions (HSIL/CIN 2-3). In this case, the vaccine should be administered early after diagnosis of the lesion, before the intervention if possible and up to 12 months thereafter.
If vaccination with more than one live attenuated vaccine is indicated, they should be administered at the same consultation in different anatomical regions. If they are not administered together, an interval of at least 4 weeks between vaccinations should be observed.
Post-vaccination serology tests should be performed one month after completion of the vaccination schedule to confirm correct response to the vaccine (anti-HBs titre >10UI/mL). In patients not showing seroconversion, vaccination should be repeated with three additional doses at monthly intervals. Revaccination should use AS04 adjuvanted vaccine or a high-load vaccine (40μg). Patients not showing seroconversion after revaccination will be considered non-responders, and post-exposure prophylaxis should be considered in situations of risk.
Vaccination processes can extend from 2 months to a year; clinicians must therefore assess the risks/benefits of vaccination. If onset of alemtuzumab treatment cannot be delayed, safety intervals should be observed between vaccination and treatment onset, as established in Table 2.
Vaccination strategy when alemtuzumab treatment is indicated.
| Vaccination before alemtuzumab | Live attenuated vaccines | Inactivated vaccines |
|---|---|---|
| No previous disease-modifying treatment | Vaccination protocol should be completed at least 6 weeks before starting alemtuzumab. | No contraindication to vaccination and no minimum interval for the start of treatment, although the response may be less than expected |
| Recent corticosteroid therapy (treatment of a relapse) | Vaccination should be started at least one month after the last dose in patients receiving prednisone >20mg/day (or equivalent) for more than 15 days.If the patient has received a corticosteroid bolus in the last three months, individual assessment of immune condition is advised. | No contraindication to vaccination, although the response may be less than expected when corticosteroids are used at immunosuppressive doses |
| Vaccination protocol should be completed at least 6 weeks before starting alemtuzumab. | ||
| Active treatment with interferon beta or glatiramer acetate | No contraindication to vaccination during treatment with these drugs (except when combined with immunosuppressants), although the response may be less than expected | |
| Vaccination protocol should be completed at least 6 weeks before starting alemtuzumab. | No contraindication to vaccination, although the response may be less than expected | |
| Active treatment with teriflunomide or dimethyl fumarate | Vaccination should be started once the washout period has been completed (Table 3).a | No contraindication to vaccination, although the response may be less than expected |
| Vaccination protocol should be completed at least 6 weeks before starting alemtuzumab. | ||
| Active treatment with specific immunosuppressors: natalizumab or fingolimod | Vaccination should be started at least three months after the last dose of any of these drugs.a | No contraindication to vaccination, although the response may be less than expected |
| Vaccination protocol should be completed at least 6 weeks before starting alemtuzumab. | ||
| Active treatment with alemtuzumab | Contraindicated during treatment. Vaccination should be administered after alemtuzumab. | No contraindication to vaccination, although the response may be less than expectedVaccination should be postponed (until after alemtuzumab) to ensure effective immunisation. |
| Vaccination after alemtuzumab | Live attenuated vaccines | Inactivated vaccines |
| Contraindicated for 6 months and until systemic immune reconstitution is achievedb | No contraindication, although the response may be less than expected. Systemic immune reconstitution should be achievedb |
DMF: dimethyl fumarate.
As the window period can be prolonged, physicians should consider starting alemtuzumab and proceed to the post-alemtuzumab vaccination section. This should not be a limitation for starting alemtuzumab, since the appropriate vaccination/immunisation schedule should have been confirmed before starting treatment with interferon beta, glatiramer acetate, teriflunomide, DMF, natalizumab or fingolimod. Each case should be assessed individually.
- •
Active infections should be excluded through clinical history taking, physical examination, and appropriate complementary tests. The presence of an active infection requires proper treatment and postponement of alemtuzumab treatment until the infection resolves.
- •
Screening for latent tuberculosis infection is recommended, preferably by means of interferon-gamma release assays and/or tuberculin test, in accordance with each hospital's protocols. Patients positive for tuberculous infection should be treated with isoniazid (300mg/day under fasting conditions, for 6 months) supplemented with vitamin B6. Treatment of the tuberculous infection should not delay the onset of treatment with alemtuzumab. Depending on the circumstances, a delay of 2-4 weeks is recommended to check the tolerability and non-toxicity of isoniazid.
- •
Screening for other infections is recommended to inform decisions about the most appropriate vaccine and the possible risk of complications (hepatitis B and C, human immunodeficiency virus, cytomegalovirus, varicella zoster, human papillomavirus, and Strongyloides in immigrants from certain endemic areas, such as Latin America and North Africa).
- •
Prevention of herpes infection: during phase III clinical trials, aciclovir showed a protective effect against these infections.13,42 Oral prophylaxis with aciclovir should be administered from the first dose of alemtuzumab (200mg/12hours for 4-8 weeks). Alternatively, valaciclovir (500mg/day) may be administered for the same period.
- •
Listeria infection risk reduction: consumption of raw or undercooked meat and unpasteurised dairy foods should be avoided for two weeks before treatment onset and for at least one month after administration or until systemic immune reconstitution, on an individual basis.
- •
After the administration of alemtuzumab, systematic use of specific preventive measures against bacterial, fungal or parasitic infections is not advised. However, in patients with suggestive symptoms, it is necessary to rule out severe infections, whether opportunistic or otherwise.
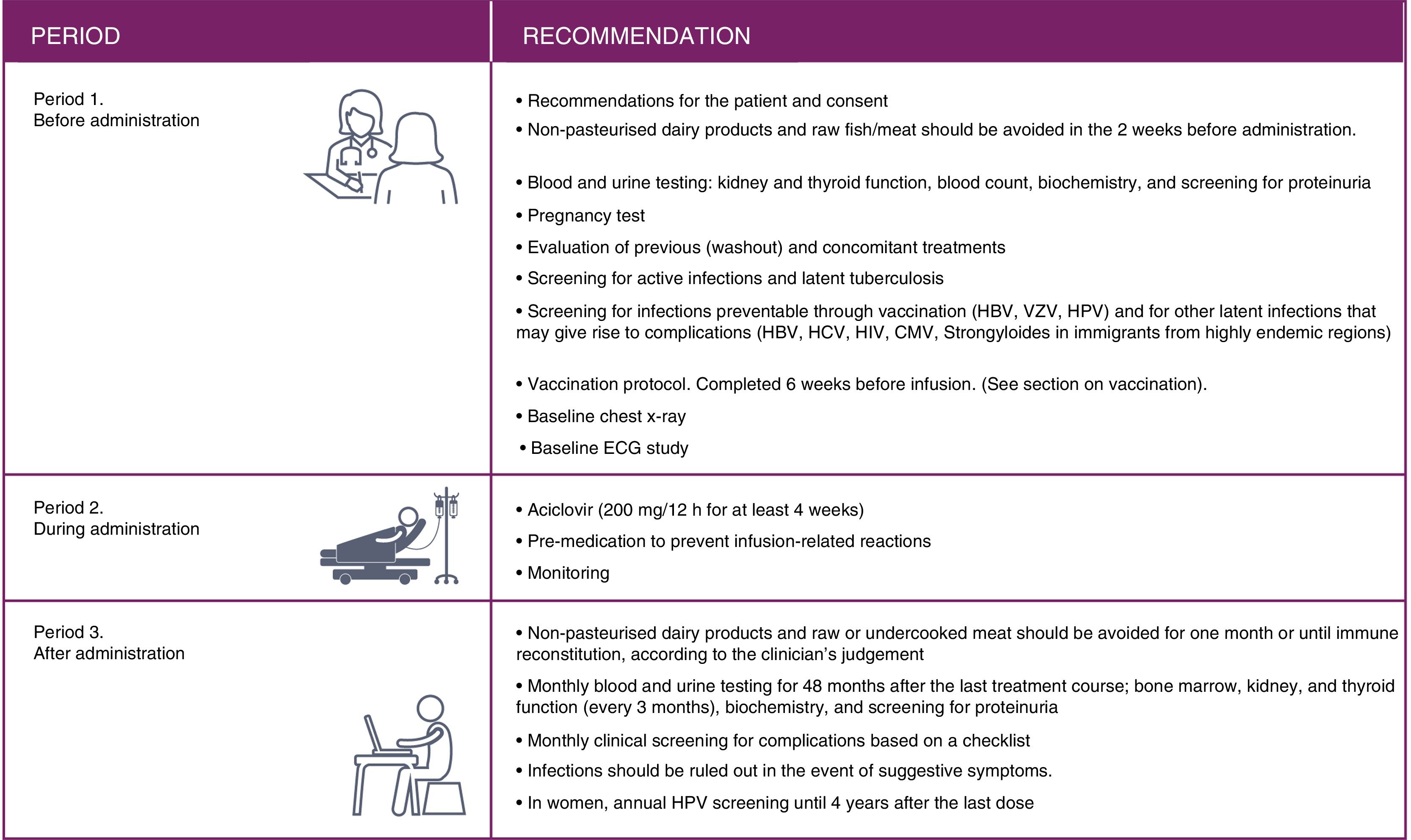
In this consensus document we differentiate three periods of alemtuzumab treatment: pre-treatment, treatment and post-treatment.
Pretreatment- •
Patients should be informed about the balance of risks and benefits of the treatment and the importance of the follow-up plan (risk minimisation plan).
- •
Dietary indications should be established for the prevention of listeria infection (see Recommendation 10).
- •
Written informed consent must be obtained.
- •
Previous (washout) and concomitant treatments should be evaluated (Table 3).
Table 3.Evaluation of previous treatments (washout) and concomitant treatments.
Other recommendations Waiting time Other recommendations Without previous treatmentInterferon betaGlatiramer acetate None – Teriflunomide – Accelerated elimination procedure 11 days after end of treatment:8g of cholestyramine orally, 3 times a day, or 50g activated charcoal orally, twice a dayBlood levels should be below 0.02mg/L.4 Dimethyl fumarate – Provided that lymphocyte count is above 800cells/μL49 Fingolimod 6 weeks Provided that lymphocyte count is above 800cells/μL50 Natalizumab 12 weeks JC (–), 12 weeks after the last infusion of natalizumab9; a gadolinium-enhanced brain MRI should be performed before perfusion of alemtuzumab to screen for PML.JC (+) the window period should allow for latent PML to be ruled out before the perfusion of alemtuzumab, although the excessive duration could favour additional relapses (also called “IRIS-like”) due to natalizumab withdrawal. In these cases, a study of the number of JC copies in the CSF should be considered to rule out latent PML.39 A gadolinium-enhanced brain MRI should also be performed to screen for PML before perfusion of alemtuzumab, and 6 months and a year after the administration of alemtuzumab.51 Other immunosuppressants (azathioprine, cyclophosphamide, rituximab) Between 3 and 6 months Provided lymphocyte count is >800cells/μL and the CD4 count >200cells/μL CSF: cerebrospinal fluid; IRIS: immune reconstitution inflammatory syndrome; JC: John Cunningham virus; MRI: magnetic resonance imaging; PML: progressive multifocal leukoencephalopathy.
- •
Baseline blood and urine analysis should be conducted before perfusion: renal and thyroid function, biochemistry, complete blood count, and proteinuria screening.
- •
Pregnancy test and contraception. Women of childbearing age should use effective contraceptive methods while receiving treatment with alemtuzumab and for 4 months after the course of treatment.15,16
- •
Screening should be performed for active, preventable, and latent tuberculosis infections. See “Questions to be considered before treatment onset.”
- •
Vaccination protocol (see “Questions to be considered before treatment onset”).
- •
Electrocardiogram and recent chest radiography are recommended. In asymptomatic patients, an X-ray performed in the 2 months prior to the day of perfusion may be valid.
The recommended dose of alemtuzumab is 12mg/day, administered by intravenous infusion in 2 initial courses of treatment, with up to 2 additional courses of treatment if needed, in accordance with the summary of product characteristics (first course: 5 days of treatment; second and posterior courses: 3 consecutive days).30
Recommendation 16- •
The administration of alemtuzumab can be performed on an inpatient or an outpatient basis. Both options are valid and the choice of which approach to follow will depend on the infrastructure available at the hospital, access to hospital beds, hospital timetables, and other factors.
- •
In accordance with the recommendations included in the section on infection risk, aciclovir (200mg/12hours) should be started before administering alemtuzumab and maintained for a minimum of 4-8 weeks.
- •
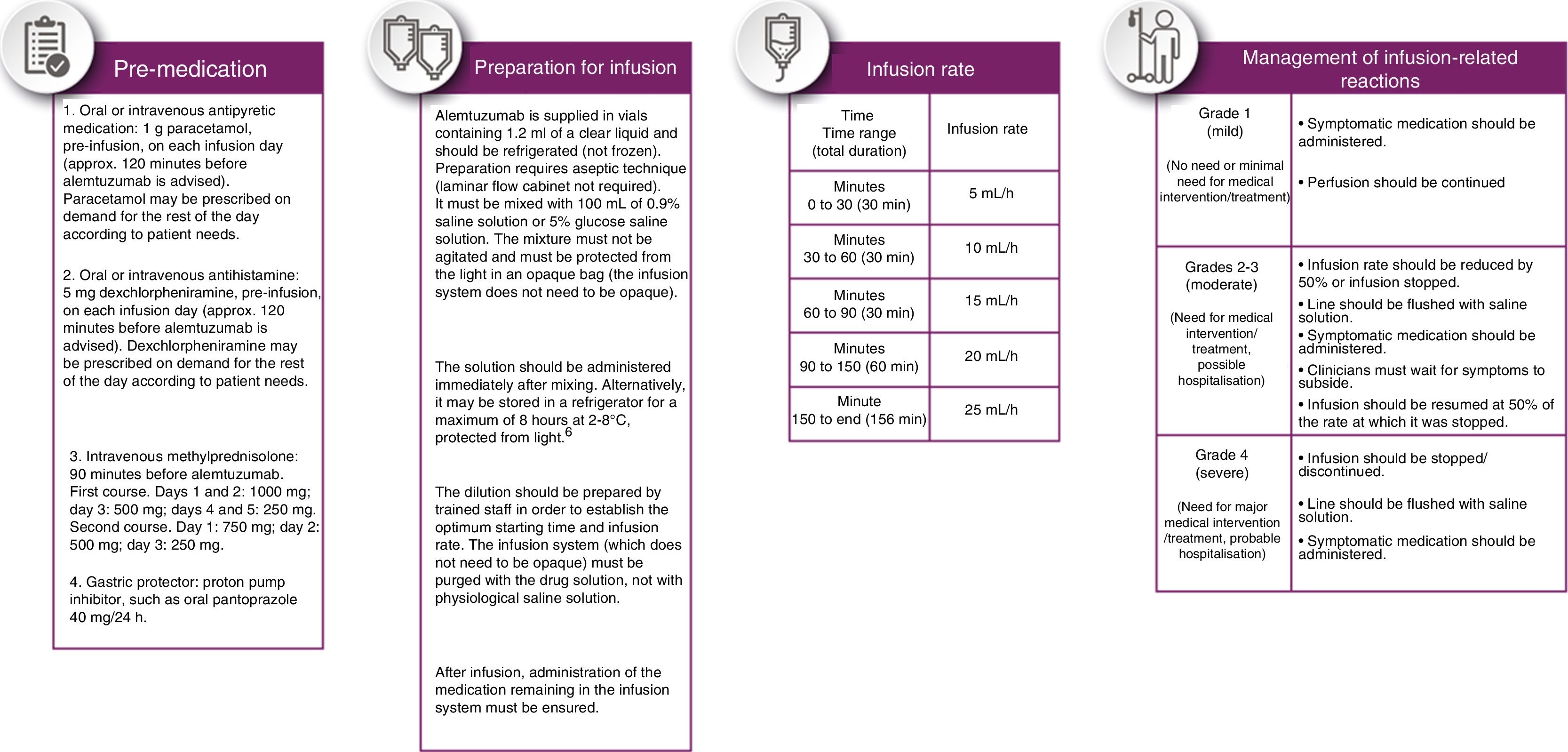
Pre-medication should be administered to reduce the incidence and intensity of infusion-related reactions (Fig. 3, pre-medication panel).

- •
The drug should be administered in a period not shorter than 4 hours or longer than 8hours; perfusion rate may be reduced if the patient presents an infusion-related reaction. The perfusion rate shown in Fig. 3 was established by consensus.
After perfusion, patients should remain under observation for 2hours, with the peripheral venous catheter remaining in place.
Monitoring during perfusion and management of infusion-related reactions- a)
Monitoring and observation during and after perfusion.
- •
Heart rate, blood pressure, and temperature should be taken every hour during infusion and during the two hours of observation after perfusion. Dermatological inspections may also be conducted to detect the presence of hives, rash, and other less frequent dermatological or vascular lesions.
- b)
Management of infusion-related reactions.
- •
Infusion-related reactions should be treated according to their severity, according to the indications included in Fig. 3.
- •
Symptomatic treatment with oral antihistamines and antipyretics may be used at home if symptoms such as rashes or headache present. Patient should be instructed to return to the hospital if symptoms persist or worsen after treatment.
- •
A patient's card with the administered medication (risk minimisation plan) should be provided.
- •
General recommendations to reduce the risk of infection: maintaining proper bodily and hand hygiene and avoiding poorly ventilated environments and places where human overcrowding occur.
- •
Dietary indications should be maintained.
- •
Blood and urine analysis should be performed monthly for 48 months after the last course: complete blood count, renal function, thyroid function (quarterly), biochemistry, and proteinuria screening.
- •
Access to other specialists (e.g., endocrinology, haematology, nephrology) should be ensured to consult about any adverse events.
- •
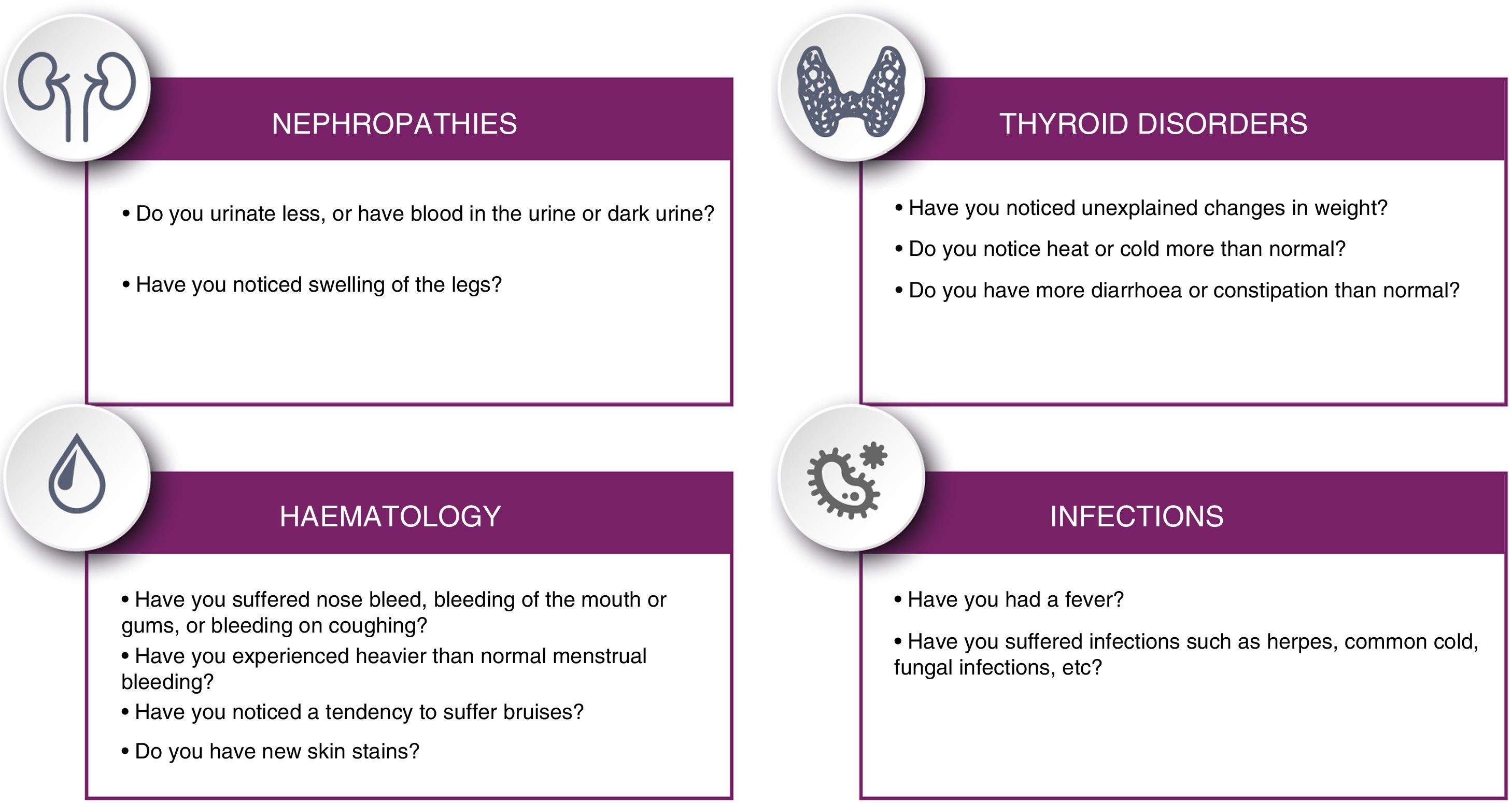
Patients should be informed of potential symptoms suggestive of immunological alterations that may present in the long term, following the checklist proposed in Fig. 4.

- •
Gynaecological cytology tests for human papillomavirus should be performed annually in women treated with alemtuzumab.
A summary of the 3 periods is included in Fig. 5.
Administration of the second and subsequent coursesRecommendation 23
- •
Twelve months must pass between the first and second courses of alemtuzumab. This period should be extended for patients not displaying systemic immune reconstitution (CD4+ T lymphocyte count >200cells/μL) or in the event of some active complication due to the first course (for example, a thyroid alteration).
In clinical trials, administration of a second course has demonstrated efficacy in patients presenting relapses after the first course.25,26 Preliminary data show that 42% of patients receiving alemtuzumab presented a significantly reduced annual relapse rate (from 1.29 to 0.34 in the first year after the second course) and did not receive additional treatment for 6 years.25,26
Recommendation 24- •
In the second and subsequent courses of alemtuzumab, the same procedures should be followed during the three established periods as in the first course.
During the CARE-MS extension studies, the administration of additional courses of alemtuzumab was permitted 12 months or more after the last course, according to the following clinical criteria: ≥ 1 relapses as defined by protocol or ≥ 2 new hyperintense lesions/increases in size of pre-existing lesions on the T2-weighted sequence, or lesions showing gadolinium uptake on the T1-weighted sequence. Decisions around whether to administer additional courses in eligible patients were taken by physicians and patients, as was the decision to start other disease-modifying therapies.15,16
An analysis of patients receiving a third course of alemtuzumab showed that the annualised relapse rate in these patients decreased from 0.74 in the year prior to this course to less than 0.1 in the following 3 years.27 Similar figures are observed in patients who did not receive an additional course. The average score on the Expanded Disability Status Scale in the 12 months after the third course decreased by 0.12, with 71% of patients scoring the same or better after this course of treatment. The safety profile in the patients receiving a third course did not change with respect to the first 2 courses.
At present, up to 2 additional courses of treatment can be administered if needed: the third or fourth course (12mg/day for 3 consecutive days) must be administered at least 12months after the previous course of treatment in patients presenting evidence of disease activity.30
Special situationsVaccination of cohabitantsRecommendation 25- •
Cohabitants and people in the immediate environment of patients treated with such immunosuppressants as alemtuzumab can be a potential source of infection. In patients with negative serology findings who have already begun treatment with alemtuzumab, it is advisable to evaluate vaccination of susceptible cohabitants against varicella and/or measles.
- •
Data on the use of alemtuzumab in pregnant women are limited. Alemtuzumab should only be administered during pregnancy if the potential benefits justify the potential risks to the foetus.30
- •
Women of childbearing age receiving alemtuzumab should use adequate contraception during the 4 months following the infusion.30
- •
It is unclear whether alemtuzumab is excreted in human breast milk. Risks for nursing infants cannot be ruled out. Therefore, breastfeeding should be interrupted during each course of treatment with alemtuzumab and for the 4 months after the last infusion of each course of treatment.30
The safety and efficacy of alemtuzumab in patients with MS between 0 and 18 years of age are yet to be established. There is no specific recommendation for alemtuzumab use in children aged between 0 and 10 years.30
Surgical interventionsRecommendation 29- •
Given the transient immunosuppression caused by alemtuzumab, it seems reasonable to delay any type of surgical intervention until adequate systemic immune reconstitution is achieved, if the clinical reason for surgery allows it. If the patient urgently requires surgical intervention during the period of severe lymphocytopaenia, the usual protocol for immunosuppressed patients should be applied.
- •
The high efficacy of alemtuzumab and its rapid implementation into MS management give rise to the need for consensus on the practical clinical management of patients receiving this treatment.
- •
This consensus statement on the use of the drug in patients with MS should not be used as guidelines for its management, but is intended to optimise and facilitate its use and follow-up in routine clinical practice.
- •
A total of 29 recommendations have been agreed upon by the panel of MS experts, and mainly cover the following aspects of treatment with alemtuzumab: characteristics of candidate patients, safety measures, vaccination of patients and cohabitants, risk of infections, previous and concomitant treatments and washout periods, contraception, complementary tests (electrocardiography and chest x-rays), cytology testing with human papillomavirus determination, infusion-related reactions, pregnancy and lactation, surgical intervention, perfusion of the drug, and the administration of successive courses after the first course of treatment.
- •
This consensus document does not address the management of possible adverse effects. This question is consulted in previous consensus documents on the management of adverse autoimmune effects with alemtuzumab.43–46
Sanofi-Genzyme provided financial support to this project.
Conflicts of interestJosé E Meca-Lallana has received consulting fees as a consultant and lecture honoraria from Biogen-Idec, Celgene, Sanofi-Genzyme, Merck-Serono, Novartis, Roche, and Teva; María Fernández-Prada has participated in teaching activities supported by Pfizer, in expert meetings promoted by Sanofi-Genzyme and GlaxoSmithKline (GSK), and collaborated in the creation of educational material for GSK; Elisa García Vázquez has received consulting fees from Sanofi-Genzyme; Santiago Moreno Guillén has received research funding and spoken at activities promoted by Abbvie, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead, GSK, Janssen Cilag, Merck Sharp & Dohme, Pfizer, Roche, and Viiv Healthcare; Susana Otero Romero has received consulting fees from Sanofi-Genzyme and lecture honoraria from Biogen-Idec and Merck Sharp & Dohme, as well as research funding from Novartis; Macarena Rus Hidalgo has received consulting fees from Novartis, Merck-Serono, Sanofi-Genzyme, Biogen-Idec, Almirall, and Bayer; Luisa M Villar Guimerans has received fees for speaking, research, and meeting and conference attendance from Merck, Biogen, Sanofi-Genzyme, Roche, and Novartis; Sara Eichau Madueño has received consulting fees and lecture honoraria from Biogen-Idec, Novartis, Sanofi-Genzyme, Merck-Serono, Roche, and Almirall; Óscar Fernández has received fees for consulting, moderating, speaking, and clinical trial and research project participation from Bayer, Biogen-Idec, Merck-Serono, Teva, Novartis, Allergan, Almirall, Sanofi-Genzyme, and Roche; Guillermo Izquierdo Ayuso has received consulting fees and lecture honoraria from Bayer, Biogen-Idec, Novartis, Sanofi-Genzyme, Merck-Serono, Almirall, Roche, Actelion, Celgene, and Teva. He has also received fees for research projects promoted by Bayer, Biogen-Idec, Novartis, Sanofi-Genzyme, Merck-Serono, Almirall, and Teva. José Carlos Álvarez Cermeño has received consulting fees from Sanofi-Genzyme, Celgene, and Merk; Carmen Arnal García has received consulting fees from Sanofi-Genzyme; Rafael Arroyo González has spoken at meetings promoted by Sanofi-Genzyme, Novartis, Teva, Roche, Biogen-Idec, Merck-Serono, and Almirall; Luis Brieva Ruíz has received research funding and honoraria for meetings promoted by Bayer, Biogen-Idec, Roche, Merck, Novartis, Allmirall, and Sanofi-Genzyme; Carmen Calles Hernández has received consulting fees from Sanofi-Genzyme; Antonio García Merino has received consulting fees and travel and meeting expenses from Bayer, Merck-Serono, Teva, Biogen-Idec, Novartis, Roche, Almirall, and Sanofi-Genzyme, and research funding from Novartis and Biogen-Idec; Montserrat González Plata has received lecture honoraria and travel expenses from Sanofi-Genzyme, Novartis, Roche, and Merck; Miguel Ángel Hernández Pérez has received consulting fees from Almirall, Biogen, Merck, Novartis, Roche, Teva, and Sanofi-Genzyme; Ester Moral Torres has received fees for consulting, speaking, and clinical trial participation from Bayer, Biogen-Idec, Merck-Serono, Teva, Novartis, Almirall, Sanofi-Genzyme, Actelion, and Roche; Javier Olascoaga Urtaza has received consulting fees from Biogen-Idec, Novartis, Roche, and Sanofi-Genzyme, and fees for moderating, speaking, and organising meetings and for involvement in research and clinical trials from Almirall, Bayer, Biogen-Idec, Merck-Serono, Novartis, Roche, Sanofi-Genzyme, and Teva; Pedro Oliva Nacarino has received consulting fees from Biogen-Idec, Bayer, and Sanofi-Genzyme, and lecture honoraria from Biogen, Novartis, Roche, Merck-Serono, Sanofi-Genzyme, Teva, and Almirall; Celia Oreja-Guevara has received consulting fees and lecture honoraria from Biogen-Idec, Merck-Serono, Novartis, Roche, Sanofi-Genzyme, and Teva; Rafa Ortiz Castillo is a medical consultant for Sanofi-Genzyme; Agustín Oterino has received lecture honoraria from Sanofi-Genzyme, Merck-Serono, Biogen-Idec, Almirall, Allergan, and Teva, and has received research funding through IDIVAL from Novartis and Sanofi-Genzyme; José María Prieto González is a consultant for Bayer, Biogen-Idec, Sanofi-Genzyme, Novartis, Sanofi-Aventis, Teva, Roche, Merck-Serono, and Almirall. He has spoken at meetings promoted by Almirall, Bayer, Biogen-Idec, Sanofi-Genzyme, Merck-Serono, Novartis, Sanofi-Aventis, and Teva. He has received research funding from Almirall, Biogen-Idec, Novartis, and Sanofi-Genzyme. Lluís Ramió-Torrentá has received consulting fees, lecture honoraria, and travel expenses from Biogen, Merck, Teva, Sanofi-Genzyme, Roche, Bayer, Almirall, and Mylan; Alfredo Rodríguez-Antigüedad has received lecture honoraria and fees for moderating and participating in clinical trials and other research projects from Bayer-Schering, Biogen-Idec, Sanofi-Genzyme, Merck, Novartis, Teva, and Roche; Albert Sáiz has received consulting fees and lecture honoraria from Bayer-Schering, Merck-Serono, Novartis, Teva, Sanofi-Genzyme, Biogen-Idec, and Roche; Mar Tintoré has received consulting fees and lecture honoraria from Almirall, Bayer, Schering, Biogen-Idec, Sanofi-Genzyme, Merck-Serono, Novartis, Roche, Sanofi-Aventis, and Teva; Xavier Montalbán Gairin has received fees for consulting in clinical trials and organising meetings from Actelion, Bayer, Biogen-Idec, Celgene, Merck-Serono, Novartis, Roche, Sanofi-Genzyme, Teva, Excemed, MSIF, and NMSS.
The authors thank Mónica Giménez for her help in drafting, reviewing, and submitting the manuscript
The following are the supplementary data to this article:
Please cite this article as: Meca-Lallana JE, Fernández-Prada M, García Vázquez E, Moreno Guillén S, Otero Romero S, Rus Hidalgo R, et al. Consenso de expertos sobre el uso de alemtuzumab en la práctica clínica diaria en España. Neurología. 2022;37:615–630.
