



Las miopatías relacionadas con el colágeno tipo vi (MCOLVI) representan un conjunto de entidades que incluyen formas más leves, como la miopatía de Bethlem (BM), y formas más graves, como la distrofia muscular congénita de Ullrich1-4. A nivel fenotípico lo que caracteriza a estas miopatías es la presencia junto a la debilidad muscular de contracturas articulares en flexión, hiperlaxitud articular de predomino distal y trastornos cutáneos (hiperqueratosis folicular, piel «aterciopelada» y cicatrices queloides). El colágeno tipo vi es una proteína estructural de la matriz extracelular constituida por 3 subunidades α, codificadas por los genes COL6A1, COL6A2 y COL6A3. A pesar de que se están describiendo constantemente nuevas mutaciones en estos genes5, establecer una correlación fenotipo-genotipo no resulta sencillo debido a la heterogeneidad clínica y al solapamiento genético6,7.
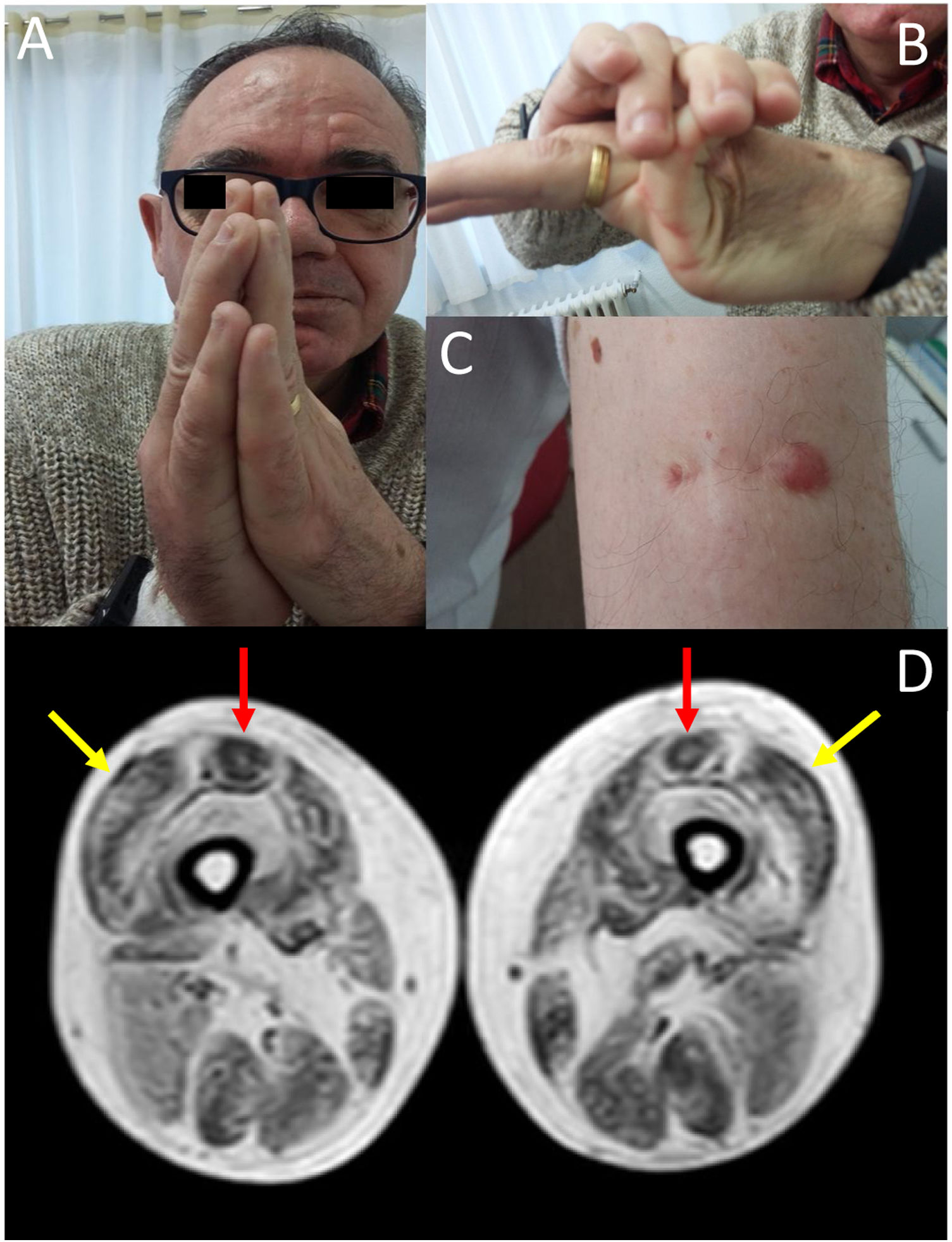
Presentamos el caso de un varón de 58 años, con diagnóstico previo de distrofia muscular de cinturas de inicio precoz, curso lentamente progresivo y con afectación respiratoria, sin defecto genético conocido. El paciente fue diagnosticado a los 2 años de edad por retraso en la adquisición en los hitos motores, describiéndose debilidad de predominio proximal y siendo la biopsia muscular compatible con distrofia muscular. El paciente mantenía debilidad proximal pero sin requerir apoyo para la marcha en el momento actual. En la exploración, junto con la debilidad 4/5 a nivel de cintura escapular y pelviana, destacaba hiperlaxitud articular de predominio en manos (fig. 1, imagen B), contracturas de las articulaciones metacarpofalángicas e interfalángicas (fig. 1, imagen A), así como cicatrices queloides en la zona de la biopsia (fig. 1, imagen C). Desde el punto de vista respiratorio, el paciente presentaba un patrón respiratorio restrictivo moderado8, sin afectación cardiológica en las pruebas complementarias. Se objetivó una elevación discreta de creatinquinasa (x2). Ante este cuadro clínico se solicitó una RM muscular que apoyase la posibilidad de MCOLVI y de esta forma dirigir en el estudio genético. La RM (ver fig. 1, imagen D) evidenció un patrón de afectación compatible con la sospecha de MCOLVI, solicitándose un estudio genético donde se identificó que el paciente era portador heterocigoto de la variante p.Gly289Val en el gen COL6A2 (secuencia de referencia del nucleótido: NM_001849.3:c.866G>T NC_000021.8:g.47535933G>T), compatible con el diagnóstico de BM. Esta variante condiciona un cambio aminoacídico, que implica a la glicina 289 de la cadena α2 del colágeno tipo vi, la cual forma parte del motivo repetitivo Gly-X-Y de la porción N-terminal del dominio triple helicoidal de la proteína, que es sustituida por el aminoácido valina. Se trataba de una mutación de tipo missense, que únicamente estaba registrada en el catálogo de variantes EmVClass como probablemente patogénica, sin que estuviese presente en otras bases de datos genéticas ni hubiera sido descrita en ninguna publicación. No obstante, las características clínicas así como las imágenes de RM muscular apoyaban el carácter patogénico de la mutación. Se realizó un estudio de segregación en los progenitores del paciente, resultando negativo lo que implicaba que la mutación se había producido de novo.
, así como de los rectos femorales, con afectación periférica y anterocentral en forma de «U» (flechas mediales, en rojo).")
En la imagen A se objetiva la presencia de contracturas articulares distales en las articulaciones metacarpofalángicas e interfalángicas. La imagen B muestra la hiperlaxitud articular del paciente. La presencia una cicatriz queloide en una zona de biopsia muscular se muestra en la imagen C. La imagen D corresponde a la RM muscular de miembros inferiores que se realizó en nuestro paciente, la cual mostró un patrón de afectación típico de las MCOLVI: degeneración grasa de los vastos laterales a nivel periférico respetando la zona central (flechas laterales, en amarillo), así como de los rectos femorales, con afectación periférica y anterocentral en forma de «U» (flechas mediales, en rojo).
Se presenta un caso de BM secundaria a una novedosa mutación del gen COL6A2, previamente no descrita como patogénica en la literatura científica (c.866G>T, p.Gly289Val). Como se ha comentado, es necesario sospechar la presencia de una MCOLVI en pacientes con debilidad muscular de predominio proximal que asocien hiperlaxitud articular distal, contracturas musculares distales y afectación cutánea (típicamente queloides). La RM muscular muestra un patrón característico9 siendo posible confirmar el diagnóstico mediante análisis genético, si bien no puede establecerse claramente una relación genotipo-fenotipo.
Desde el punto genético, clásicamente se ha descrito que la distrofia muscular congénita de Ullrich se heredaba de forma AR y la BM de forma AD, si bien ambos patrones de herencia se han sido descritos para todo el espectro de patologías relacionadas con el colágeno tipo vi5,10-13. Mediante secuenciación genética es posible realizar un análisis mutacional de los exones de los genes COL6A1, COL6A2 y COL6A3. Las variantes de tipo missense, como la hallada en nuestro paciente, suelen aparecer de novo en heterocigosis, con un efecto dominante-negativo. En concreto, las variantes de tipo missense que afectan a los dominios repetitivos Gly-X-Y de la porción N-terminal del dominio triple helicoidal del colágeno tipo vi son la causa más frecuente de las patologías relacionadas con dicho colágeno11. Por último, cabe destacar que se han descrito otras 3 variantes patogénicas que afectan al mismo residuo aminoacídico, c.865G>T, p.Gly289Arg14; c.865G>T, p.Gly289Cys14; c.866G>A, p.Gly289Asp (esta última presente en la base de datos mutacional de ClinVar).
Conflicto de interesesTodos los autores declaran no tener conflictos de interés en la publicación de este artículo.

