



Collagen VI (ColVI)–related myopathies encompass a group of diseases ranging from milder muscle disorders, such as Bethlem myopathy, to more severe forms, such as Ullrich congenital muscular dystrophy.1–4 From a phenotypic viewpoint, these disorders are characterised by muscle weakness, joint contractures in flexion, joint hypermobility predominantly affecting distal joints, and skin alterations (follicular hyperkeratosis, “velvety” skin, and keloid scars). Collagen VI is a structural protein of the extracellular matrix, comprising 3 subunits, encoded by the genes COL6A1, COL6A2, and COL6A3. Although new mutations in these genes are constantly being reported,5 establishing a genotype-phenotype correlation is challenging due to the clinical heterogeneity and genetic overlaps.6,7
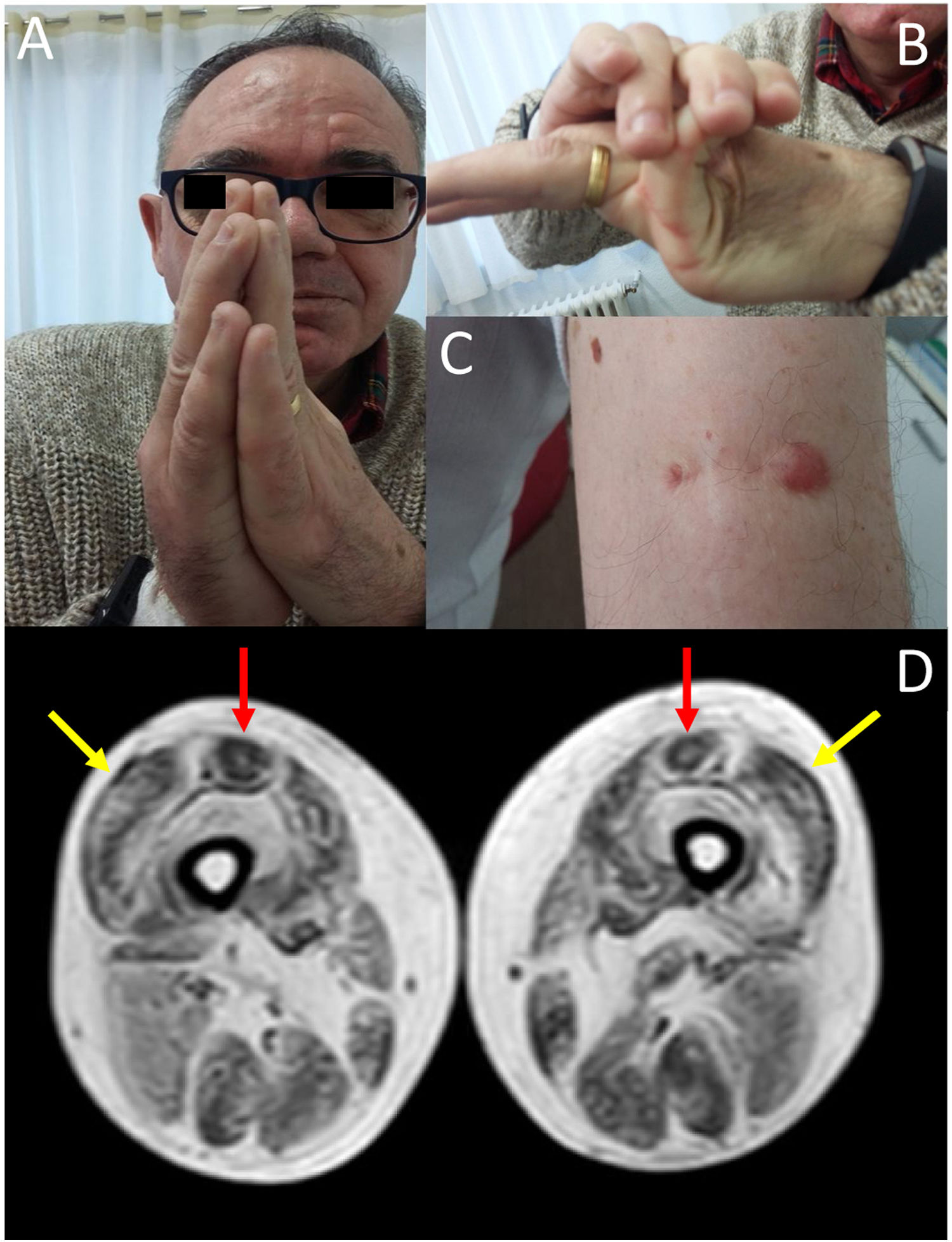
We present the case of a 58-year-old man with history of early-onset, slowly progressing limb-girdle muscular dystrophy associated with respiratory involvement and no known genetic defects. Diagnosis had been established when the patient was 2 years old due to delayed acquisition of developmental milestones; weakness predominantly affected distal muscles, and a muscle biopsy revealed signs compatible with muscular dystrophy. At the time of assessment at our department, the patient presented proximal muscle weakness but did not require support to walk. The examination revealed 4/5 muscle strength at the level of the shoulder and pelvic girdles, joint hypermobility predominantly in the hands (Fig. 1B), contractures in the metacarpophalangeal and interphalangeal joints (Fig. 1A), and keloid scars in the area where the muscle biopsy specimen was taken (Fig. 1C). From a respiratory viewpoint, the patient presented a pattern of moderate restrictive pulmonary function,8 with complementary tests revealing no cardiac alterations. He also presented increased creatine kinase levels (×2). A muscle MRI scan was performed to confirm suspicion of ColVI-related myopathy and to guide the genetic study. The MRI scan (Fig. 1D) showed a pattern compatible with ColVI-related myopathy, and a genetic study revealed that the patient was a heterozygous carrier of variant p.Gly289Val in the gene COL6A2 (reference sequence: NM_001849.3:c.866G>T NC_000021.8:g.47535933G>T); this finding is compatible with diagnosis of Bethlem myopathy. This mutation causes an amino acid change involving glycine at position 289 of the α2 chain of collagen VI, which forms part of the Gly-X-Y motif in the N-terminal region of the triple helical domain of the protein, with this amino acid being substituted by valine. This missense mutation was classified as probably pathogenic in the EmVClass catalogue, but had not been listed in any other genetic database, nor had it been reported in previous publications. However, our patient’s clinical characteristics and muscle MRI findings support the pathogenic nature of the mutation. A family segregation study did not identify the mutation in our patient’s parents, meaning that the mutation was de novo.
 Contractures affecting the metacarpophalangeal and interphalangeal joints. (B) Joint hypermobility. (C) Keloid scar in the area where a muscle biopsy specimen was taken. (D) Lower-limb MRI scan showing a pattern compatible with ColVI-related myopathy: fatty degeneration in the periphery of the vastus lateralis muscles (yellow arrows) and in the periphery and anterior-central area of the rectus femoris muscles, with a U-shaped pattern (red arrows).")
(A) Contractures affecting the metacarpophalangeal and interphalangeal joints. (B) Joint hypermobility. (C) Keloid scar in the area where a muscle biopsy specimen was taken. (D) Lower-limb MRI scan showing a pattern compatible with ColVI-related myopathy: fatty degeneration in the periphery of the vastus lateralis muscles (yellow arrows) and in the periphery and anterior-central area of the rectus femoris muscles, with a U-shaped pattern (red arrows).
We present a case of Bethlem myopathy secondary to a novel COL6A2 mutation (c.866G>T, p.Gly289Val); this mutation had not previously been described as pathogenic in the literature. ColVI-related myopathy should be suspected in patients with predominantly proximal muscle weakness and distal joint hypermobility, distal muscle contractures, and skin alterations (typically keloids). Muscle MRI reveals a characteristic pattern,9 and diagnosis can be confirmed with genetic studies, although a clear genotype-phenotype correlation cannot be established.
From a genetic viewpoint, Ullrich congenital muscular dystrophy has typically been reported to follow an autosomal recessive inheritance pattern, whereas Bethlem myopathy is thought to follow an autosomal dominant pattern. However, both inheritance patterns have been described in the spectrum of ColVI-related myopathies.5,10–13 Genetic sequencing enables analysis of exonic mutations in COL6A1, COL6A2, and COL6A3. Missense mutations, like the one identified in our patient, usually appear de novo in heterozygosis, exerting a dominant-negative effect. More specifically, missense mutations in the Gly-X-Y motif in the N-terminal region of the triple helical domain of collagen VI constitute the most frequent cause of ColVI-related myopathies.11 Lastly, 3 other pathogenic variants are reported to involve the same amino acid residue: c.865G>T, p.Gly289Arg14; c.865G>T, p.Gly289Cys14; and c.866G>A, p.Gly289Asp (the latter is included in the ClinVar database).
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Martínez-Martín Á, Díaz-Maroto Cicuéndez I, Simón Sánchez J, García-García J. Debilidad muscular, laxitud articular y queloides. Una asociación más que sugerente. Neurología. 2021;36:243–245.
