



Gammahydroxibutiric aciduria is a rare condition caused by a deficiency of succinic semialdehyde dehydrogenase, an enzyme involved in gamma-aminobutyric acid (GABA) degradation. The condition manifests with non-specific neurological symptoms, and no effective treatment is currently available.
We present the case of a 5-year-old boy with no relevant medical history who visited our centre's neuropaediatric unit due to psychomotor delay. He had presented delay in achieving motor development milestones: head control at 6 months, sitting up at 9 months, and walking at 24 months. The patient's parents reported that he said words like “mama” and “dada” when he was one. The first examination of the patient was conducted when he was 11 months old, finding hypotonia with hypoactive reflexes in the lower limbs. No alterations were observed in the electromyography or laboratory analysis, which included CPK and thyroid hormone measurements. He was subsequently followed up in another hospital; karyotyping, fragile X study, electroencephalography (EEG), and auditory evoked potential study yielded normal results.
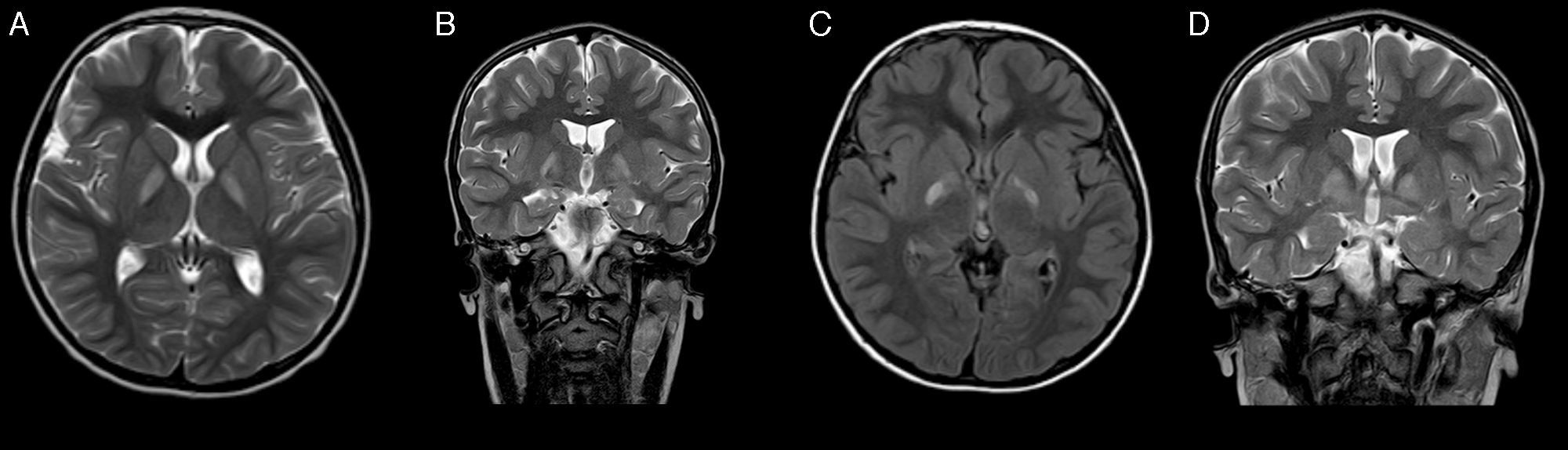
At the age of 4 years, he returned to our department. The most remarkable finding was a developmental delay; this was most pronounced in expressive language and communication, which was predominantly symbolic. A new EEG and otorhinolaryngological examination again showed normal results. He subsequently presented oppositional-type behavioural changes and hand stereotypic movements. A magnetic resonance imaging (MRI) scan revealed bilateral, selective, and symmetrical involvement of the globus pallidi (Fig. 1). We requested blood and urine metabolic tests, which detected increased urinary excretion of 4-hydroxybutyric acid and threo- and erythro-4,5-dihydroxyhexanoic acids. As succinic semialdehyde dehydrogenase deficiency was suspected, the ALDH5A1 gene was studied and 2 known heterozygous mutations were found: p.Trp204Ter (c.612G>A) and p.Gly268Glu (c.803G>A). Genetic study of the patient's parents confirmed that they were carriers of these mutations. We suggested treatment with vigabatrine, which was rejected by the parents.
 Axial T2-weighted sequence; (B) coronal T2-weighted sequence; (C) axial FLAIR sequence at 2 months after A and B; (D) coronal T2-weighted sequence (2 months after A and B).")
Several months after diagnosis, the patient experienced sudden-onset weakness in the left limbs, predominantly affecting the arm, which showed impaired motor function; this progressed to dystonic hemiparesis after 4 days. The patient was receiving risperidone; he also manifested fatigue and episodes of disorientation lasting several seconds, with no associated abnormal movements. A brain CT scan showed no significant changes with regards to the previous CT scan, and an EEG showed synchronous and independent focal epileptiform activity in both temporal regions, predominantly on the right side. Treatment with risperidone was discontinued; clinical symptoms did not improve and behavioural alterations worsened, so treatment with lamotrigine was started. A subsequent MRI showed a favourable evolution of the previous lesions to the globus pallidi, revealing no other findings (Fig. 1). Dystonia improved but a moderate distal involvement persisted; episodes of disorientation resolved with antiepileptic treatment.
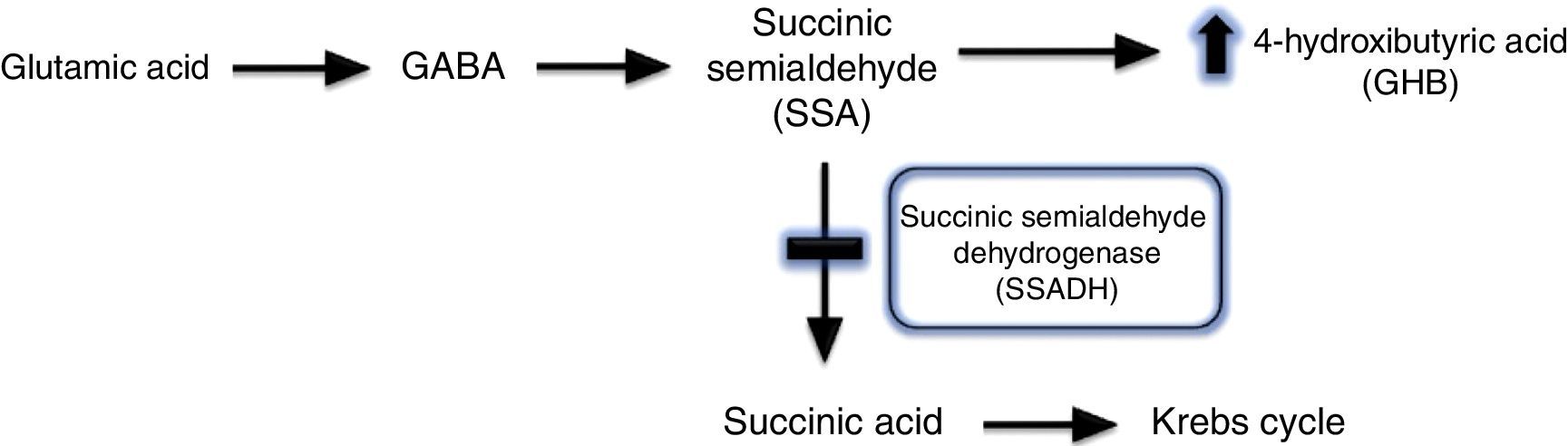
Gammahydroxibutiric aciduria was first described in 1893 and is caused by succinic semialdehyde dehydrogenase (SSADH) deficiency. A transamination process converts GABA into succinic semialdehyde, which is in turn degraded into succinic acid by the action of SSADH. SSADH deficiency involves an increase in levels of the potentially neurotoxic 4-hydroxybutyric acid (GHB) in physiological fluids (Fig. 2).1,2 This is a metabolic disease with an autosomal recessive inheritance pattern, which may be originated by different mutations of the ALDH5A1 gene, located on the short arm of chromosome 6.3 The true prevalence of this condition remains unknown: 350 cases have been published in the literature to date, the majority in paediatric patients.

These cases show differing, non-specific clinical manifestations, with such neurological symptoms as hypotonia, hyporeflexia, non-progressive ataxia, and psychomotor delay, predominantly in language.4 Up to 50% of these patients present seizures throughout the course of the disease; these are usually refractory to medical treatment.2 Psychiatric alterations, which are the most incapacitating, are described in most cases. These range from autistic features to hallucinations, aggressiveness, hyperactivity, and sleep disorders. Mean age at diagnosis is 6.6 years (range, 0-25 years).5
Electrophysiological studies may yield normal results in nearly half of patients, and the most frequent alterations in the case series published are usually focal and generalised epileptiform activity, and slowing of background activity.2 The neuroimaging technique of choice for diagnosis is MRI, which characteristically shows a bilateral, symmetrical hyperintensity in the globus pallidi on T2-weighted sequences. Other structural findings include a predominantly vermian cerebro-cerebellar atrophy, and cortical, cerebellar, and dentate nucleus hyperintensities.2,4,6 Spectroscopy studies show increased levels of GABA and GHB in white and grey matter.7
To establish a diagnosis of SSADH deficiency, mass spectrometry or gas chromatography are used to analyse the presence of GHB or GHB isomers in urine, plasma, or cerebrospinal fluid.8 Diagnosis is confirmed through the quantification of SSADH enzyme activity in leukocytes,9 or molecular genetic study of the ALDH5A1 gene.3 In families in which the condition is detected, prenatal diagnosis is possible by DNA analysis or GHB determination in amniotic fluid or chorionic villus sampling.10 Some authors have proposed that quantification of GHB in dried blood spots during the neonatal period may help establish early diagnosis of the disease.11
Treatment options are limited; although multiple alternatives have been tested, none have been shown to be effective in all cases. Vigabatrine is a GABA-transaminase inhibitor; low doses (25mg/kg/day) decrease GHB levels in cerebrospinal fluid, with variable effects. Authors including Howells et al.12 believe that this inconsistency in the results may be due to the fact that this inhibition does not exist at the peripheral level. However, this treatment also has adverse effects, with decreased visual field, occasionally irreversible, being the most frequent. Studies with other drugs have not yet provided promising results; these studies address taurine, GHB receptor antagonist, NCS-382, and the ketogenic diet, among other treatments.13
Furthermore, treatment of concomitant symptoms should be started; these symptoms include seizures, to be treated mainly with lamotrigine and carbamazepine,14 and behavioural disorders, to be treated with methylphenidate, risperidone, and benzodiazepines.15
Management of these patients must be multidisciplinary, involving such non-pharmacological measures as rehabilitation, speech therapy, and psychological and occupational therapy.
Although SSADH deficiency is a rare disease, it should be considered in the differential diagnosis of patients with psychomotor delay, alterations in language development, movement disorders such as ataxia, hypotonia, and behavioural disorders. Epilepsy may manifest over the course of the disease, but dystonic hemiparesis, as observed in our patient, is not frequent.
FundingThe authors have received no funding of any kind.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Manrique Martín G, Ferrero García-Loygorri C, Jiménez Domingo A, Miranda Herrero MC. Retraso psicomotor, hipotonía y alteraciones del comportamiento: un caso de deficiencia de succínico semialdehído deshidrogenasa. Neurología. 2018;33:63–65.
