



Tay-Sachs disease is an autosomal recessive neurodegenerative disorder characterised by a mutation or deletion of the hexosaminidase A gene (HEXA), located at 15q23. The gene codes for the alpha subunit of hexosaminidase A, a lysosomal enzyme involved in ganglioside metabolism.1,2
Tay-Sachs disease is a lysosomal storage disease and generally manifests after a period of normal neurological development. In this disease, gangliosides (GM2), a type of sphingolipid present in the membranes of cells of the central nervous system, are stored due to the lack of hexosaminidase A. The accumulation of gangliosides causes irreversible neurological damage and death at young ages.3,4
We present the case of a 5-year-old girl with clinical, biochemical, and molecular symptoms of Tay-Sachs disease.
Our patient was born in Colombia, and was the mother's second child; the older sibling was healthy. No alterations were observed during her gestation, except for the mother's subjective perception of reduced fetal movement. When she was 8 days old, she presented frequent vomiting with no food content and mobilisation of secretions which became persistent; no further relevant pathological findings were observed until 9 months of age, when initially distal and subsequently generalised myoclonic seizures appeared.
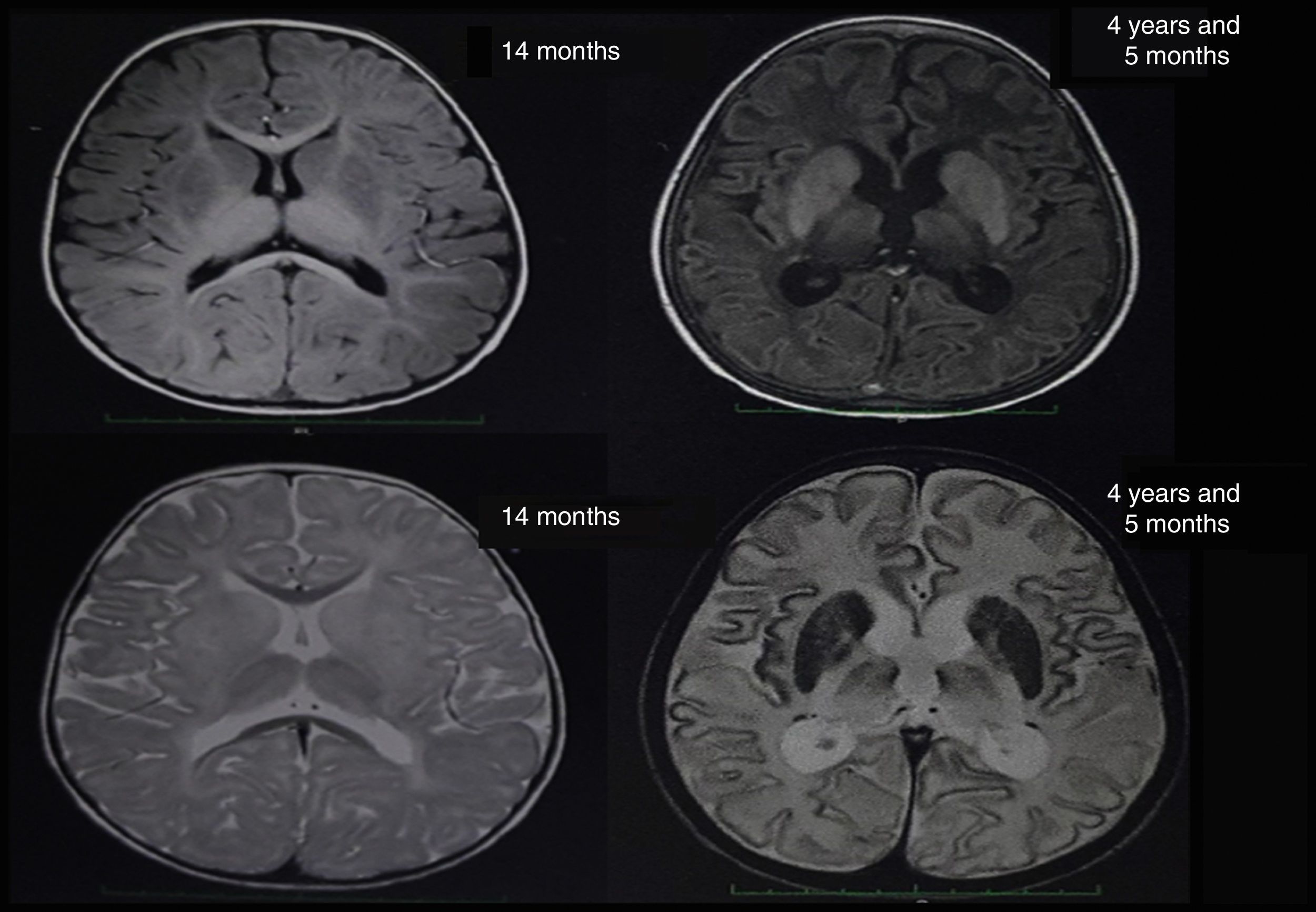
At about 10 months of age, the patient showed some regression in developmental milestones: she did not follow objects with her eyes or grasp objects and had a weak suck. At 11 months of age, she suffered a tonic-clonic seizure which led her to convulsive status epilepticus and was transferred to an emergency department; a brain CT scan showed no alterations. A brain MRI scan was performed when she was 14 months old (Fig. 1), revealing alterations in the myelination pattern, which affected the basal ganglia (mainly the putamen, globus pallidus, and caudate nucleus). A discrete component was observed in the posterior thalami and corticospinal tracts at the level of the midbrain. These findings were compatible with hypomyelination.

From the age of 11 months, our patient began to display progressive neurological impairment and recurrent aspiration pneumonia, which led to the performance of a gastrostomy to decrease episodes of pulmonary aspiration. At the age of 3.5 years, she became completely disconnected from her environment, and hypotonia progressed to generalised spasticity; when the patient was 4 years of age, she presented central apnoea and underwent a tracheostomy connection to a continuous positive airway pressure system.
In the light of the clinical symptoms described, Tay-Sachs disease was suspected and a quantification of enzymatic activity of hexosaminidase in serum yielded normal levels; however, hexosaminidase A levels were critically diminished at 5.88% (reference values between 58% and 68%). The presence of lipids in neuronal cells was confirmed by magnetic resonance spectroscopy. Significantly reduced levels of N-acetyl aspartate and preserved levels of choline were observed (loss of N-acetyl aspartate is typical of demyelinating diseases). The brain nuclear MRI scan revealed greater hyperintensities in the thalami and involutionary changes secondary to the disease in comparison to the MRI scan performed at 14 months of age (Fig. 1). Given the laboratory test results, sequencing studies of the HEXA gene were requested and revealed a heterozygous pathogenic mutation of intron 9, with a change in the nucleotide c. 1073+1G>A. This mutation has previously been reported as pathological by Akli et al.5 in 1991.
Tay-Sachs disease, described in 1881 by Tay and in 1887 by Sachs, is currently one of the most widely studied sphingolipidoses within the context of lysosomal storage diseases.6 The characteristic inheritance pattern of this disease, as in most inborn errors of metabolism, follows an autosomal recessive inheritance pattern. Mutation of the HEXA gene is responsible for the disease.2
Symptom onset occurs at 3-6 months of age; in our patient, the disease initially manifested as generalised tonic-clonic seizures at the age of 11 months. She later displayed progressive loss of the motor functions acquired until then; a significant swallowing disorder, generally leading to gastrostomy for total enteral nutrition; generalised spasticity; and invasive mechanical ventilation.7 Blindness due to optic atrophy, diagnosed at 2 years of age in our case, is another common pathological finding in this disease.8
One of the ophthalmological signs described in this disease are the cherry-red spots at the macula which correspond to the abnormal accumulation of metabolic products inside the retinal ganglion cells.9 However, in this particular case, these spots were not present, although the physical examination of the eye did reveal alterations in the fovea in a “bull's eye” shape, which may be secondary to drug-related toxicity or optical degeneration.10
A diagnosis of Tay-Sachs disease should be suspected upon onset of early symptoms of the disease, which range from an exaggerated acoustic startle reflex to the presence of psychomotor retardation and loss of previously acquired motor abilities, as in our patient. However, definitive diagnosis is established by the quantification of hexosaminidase A activity,11 which was markedly diminished in our case. Additionally, sequencing of the HEXA gene is recommended: results from our patient were positive for a single heterozygous mutation, which implies the presence of another mutation which was not identified by the molecular diagnostic methods used.
Prevalence of the disease is estimated at 1 case per 200000 live births in the general population.12 However, there is a high prevalence among Ashkenazi Jews, at approximately 1 case per 4000 live births.13
Finally, Tay-Sachs disease must be included in the differential diagnosis of paediatric neurological disorders causing regression of developmental milestones. It is important to include molecular testing as part of diagnostic protocols for the disease and genetic counselling as a means to prevent familial recurrences of the disease.
Conflicts of interestThis study has not been published in any other journal nor has it been submitted for publication to any other journal. All authors cited have reviewed this manuscript and endorsed its publication. The authors have no conflicts of interest to declare.
This study was financed by ICESI University.
Please cite this article as: Posso Gomez LJP, Gomez JF, Botero V, Pachajoa H. Caracterización clínica, bioquímica y molecular de una paciente colombiana con enfermedad de Tay-Sachs. Neurología. 2018;33:61–63.
