



La enfermedad de Tay-Sachs es un trastorno neurodegenerativo, autosómico recesivo, que se caracteriza por una mutación o deleción en el gen de la hexosaminidasa A (HEXA), ubicado en la posición 15q23; este gen codifica para la subunidad alfa de hexosaminidasa A, una enzima lisosomal que participa en el metabolismo de los gangliósidos1,2.
La enfermedad de Tay-Sachs está incluida dentro de las enfermedades de depósito lisosomal y se manifiesta generalmente después de un periodo de desarrollo neurológico normal, donde los gangliósidos (GM2), un tipo de esfingolípidos de la membrana de células del sistema nervioso central, se almacenan a causa de la carencia de la hexosaminidasa A. La acumulación de gangliósidos genera daño neurológico irreversible y la muerte del paciente a edades tempranas3,4.
Presentamos el caso de una niña de 5 años, con enfermedad de Tay-Sachs caracterizada clínica, bioquímica y molecularmente.
El caso corresponde a una paciente de 5 años de edad procedente de Colombia, producto del segundo embarazo, primer hijo sin patología. Embarazo que cursó sin alteraciones, salvo percepción subjetiva de la madre de pocos movimientos fetales. A partir de los 8 días de vida, presentó emesis frecuentes sin contenido alimentario y movilización de secreciones que se volvió persistente; no hubo más hallazgos patológicos relevantes hasta los 9 meses de edad, tiempo en el cual aparecieron mioclonías inicialmente distales y posteriormente generalizadas.
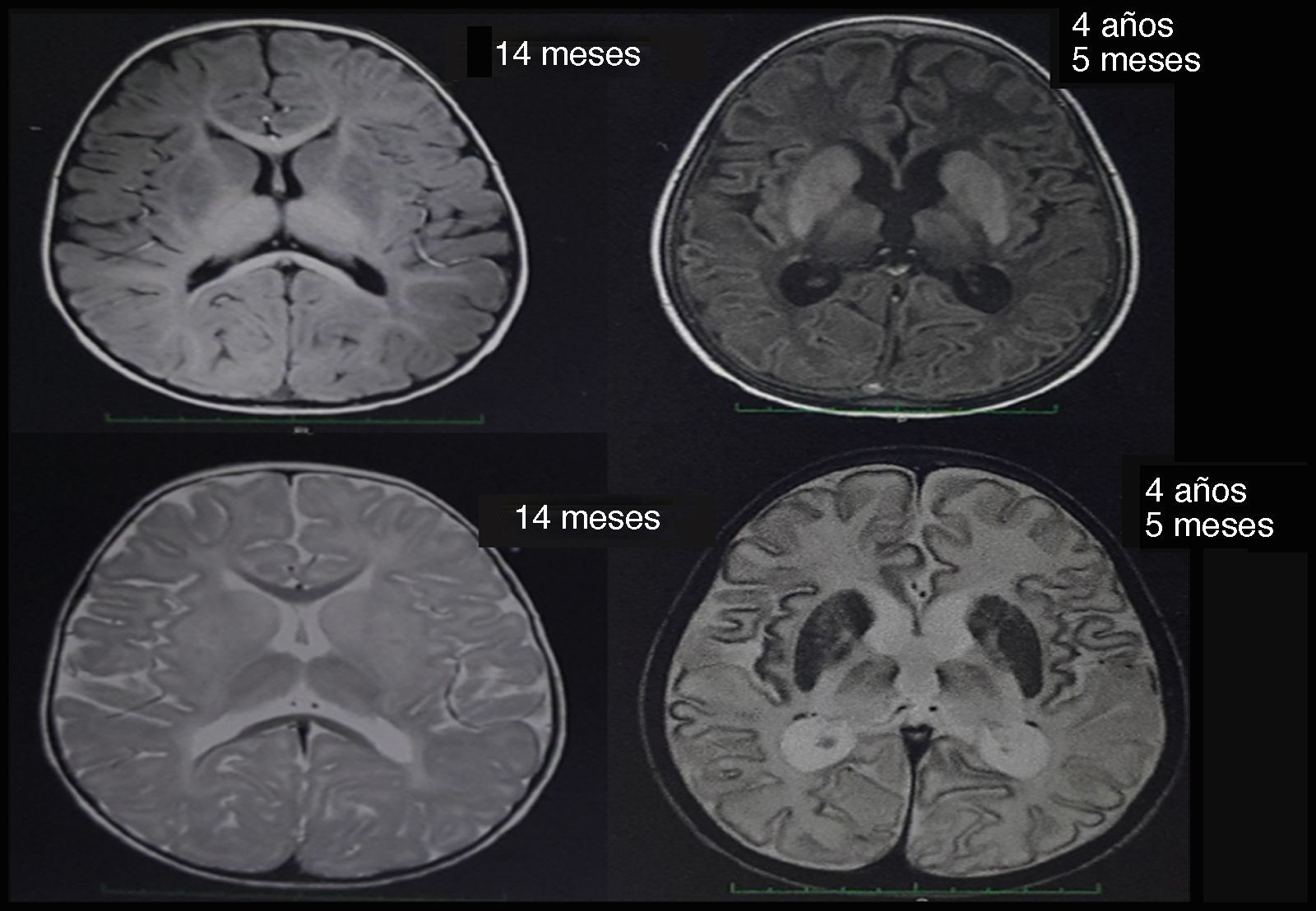
Alrededor de los 10 meses, presenta involución de los hitos del desarrollo, no seguía los objetos con la mirada, no tomaba los objetos con la mano y la succión del pecho era débil. A los 11 meses de vida la paciente experimentó convulsión tónico-clónica, llegando hasta el estatus convulsivo, por lo que fue trasladada a un servicio de urgencias donde se le realizó tomografía cerebral que no mostró ninguna alteración en ese momento, por lo que se le practicó una resonancia magnética nuclear cerebral a los 14 meses de edad (fig. 1), donde se observó alteración del patrón de mielinización, involucrando ganglios basales, principalmente putamen, globo pálido y caudado, con un componente discreto en topografía de aspecto posterior de tálamos y en topografía de tractos córtico-espinales a nivel del mesencéfalo, siendo compatible con hipomielinización.

Desde los 11 meses de edad la paciente experimentó un deterioro neurológico progresivo, neumonías aspirativas a repetición, razón por la cual decidieron realizarle una gastrostomía para disminuir los episodios broncoaspirativos. A los 3 años y medio, presenta pérdida total de la conexión con el entorno y pasó de la hipotonía a la espasticidad generalizada; a los 4 años presentó apneas centrales, por lo que se le practicó una traqueostomía con conexión a un sistema de presión positiva continua en la vía aérea.
De acuerdo con el cuadro clínico descrito, se sospechó enfermedad de Tay-Sachs, por lo que se cuantificó la actividad enzimática de la hexosaminidasa total en suero, reportada como normal, y además niveles de hexosaminidasa A, los cuales se encontraban críticamente disminuidos, 5,88%(con valores de referencia entre el 58 y el 68%); se demostró la presencia de lípidos en las células neuronales, a través de espectroscopia por resonancia magnética, encontrándose además reducciones importantes de N-acetil aspartato, niveles de colina conservados (la pérdida de N-acetil aspartato suele verse en patologías de tipo desmielinizante) y hallazgos en la resonancia magnética nuclear cerebral, encontrándose una mayor hiperintensidad de los tálamos y cambios involutivos secundarios a la enfermedad en comparación con la resonancia de los 14 meses (fig. 1). De acuerdo con los resultados de laboratorio arrojados, se solicitó secuenciación del gen HEXA, reportándose una mutación heterocigota patogénica en el intrón 9, con un cambio en el nucleótido c.1073+1G>A, mutación previamente reportada como patológica por Akli et al. en 19915.
La enfermedad de Tay-Sachs, descrita en 1881 por Tay y en 1887 por Sachs, es una de las esfingolipidosis más estudiadas hasta el momento dentro del contexto de las enfermedades por depósito lisosomal6. El patrón de herencia característico de este trastorno, como la mayoría de los errores innatos del metabolismo, es autosómico recesivo. La mutación en el gen de la HEXA es la responsable de la patología2.
El inicio de los síntomas se da alrededor de los 3 a 6 meses; en nuestra paciente, la enfermedad comenzó con convulsiones tónico-clónicas generalizadas a la edad de 11 meses; posteriormente, pérdida progresiva de las funciones motoras adquiridas hasta ese momento, trastorno de la deglución importante que lleva generalmente a la nutrición enteral total por gastrostomía, espasticidad generalizada y ventilación mecánica invasiva;7 además de ceguera producto de la atrofia óptica, como en este caso diagnosticada a los 2 años de edad, siendo este uno de los hallazgos patológicos comunes de la enfermedad8.
Uno de los signos oftalmológicos descritos en este trastorno son los puntos rojo-cereza en la mácula, que corresponden a la acumulación anormal de productos metabólicos dentro de las células ganglionares de la retina9; sin embargo, en este caso particular, estos puntos no estaban presentes en la paciente; en el examen físico ocular se halló alteración de la fóvea con aspecto de «ojo de buey», la cual puede ser secundaria a toxicidad por medicamentos o la degeneración óptica10.
El diagnóstico de la enfermedad de Tay-Sachs debe sospecharse con el inicio de los síntomas tempranos de la enfermedad, que van desde una respuesta exagerada al sonido, hasta la aparición de retraso psicomotor y pérdida de capacidades motrices adquiridas previamente, como ocurrió con nuestra paciente. Sin embargo, el diagnóstico definitivo se lleva a cabo mediante la cuantificación de la actividad enzimática de la hexosaminidasa A11, en este caso notoriamente disminuida; adicionalmente, se recomienda hacer secuenciación del gen HEXA, que en la paciente arrojó un resultado positivo para una sola mutación heterocigota, lo que supone la presencia de otra mutación no identificada por los métodos de diagnóstico moleculares utilizados.
La prevalencia estimada de la enfermedad en la población general es de 1 en 200.000 nacidos vivos12. Sin embargo, existe una alta prevalencia entre los judíos askenazíes, aproximadamente 1 de cada 4.000 nacidos vivos13.
Finalmente, la enfermedad de Tay-Sachs debe considerarse como diagnóstico diferencial de las enfermedades neurológicas infantiles que causen regresión de los hitos del desarrollo; es importante tener en cuenta la utilización de pruebas moleculares como parte de los protocolos de diagnóstico de la enfermedad y el uso de asesoría genética como herramienta para prever recurrencias familiares de la enfermedad.
Conflicto de interesesPor medio de este documento, declaro que este material no ha sido publicado previamente ni se encuentra sometido para ser publicado en otra revista; certifico que este manuscrito ha sido revisado por todos los autores citados y avalado para su publicación. También declaro no poseer conflictos de interés sobre el presente artículo.
La realización de este trabajo ha sido financiada por la universidad ICESI.

