



La aciduria gammahidroxibutírica es una enfermedad rara, autosómica recesiva, producida por el déficit del enzima succínico semialdehído deshidrogenasa, implicado en la degradación del ácido gamma-aminobutírico (GABA). La afectación es neurológica, con sintomatología inespecífica, sin existir actualmente ningún tratamiento eficaz.
Presentamos el caso de un niño de 5 años sin antecedentes de interés, seguido en la consulta de neuropediatría por retraso psicomotor. Había presentado retraso en la adquisición de hitos motores, sostén cefálico a los 6 meses, sedestación a los 9 meses y deambulación autónoma a los 24 meses. Referían emisión de primeros bisílabos referenciales al año. Valorado inicialmente a los 11 meses, objetivándose en la exploración hipotonía con reflejos hipoactivos en miembros inferiores. Se realizaron electromiograma y estudio analítico, incluyendo CPK y hormonas tiroideas, sin alteraciones. Posteriormente fue seguido en otro centro hospitalario, donde se hizo cariotipo y estudio de X frágil, electroencefalograma (EEG) y potenciales evocados auditivos dentro de la normalidad.
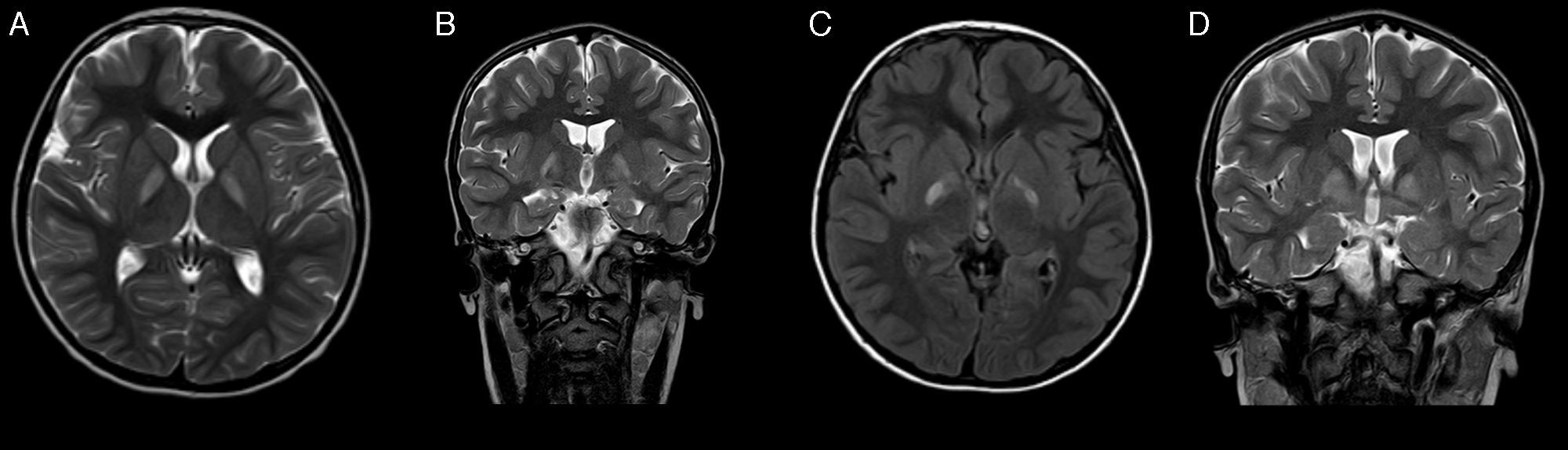
A los 4 años de vida acude de nuevo a nuestra consulta. Destacaba un retraso madurativo, más acusado en el área del lenguaje expresivo y en la comunicación, que era predominantemente simbólica. Se repitieron el EEG y la evaluación por otorrinolaringología, nuevamente sin hallazgos. Posteriormente presenta alteraciones de la conducta tipo oposicionista y estereotipias motoras de manos. En la resonancia magnética (RM) (fig. 1) realizada se objetivaba afectación selectiva bilateral y simétrica de núcleos pálidos. Por ello se solicitó estudio metabólico en sangre y orina, encontrándose excreción aumentada de ácido 4-OH-butírico y de las lactonas treo y eritro del ácido 4,5-dihidroxi-hexanoico en orina. Ante la sospecha de deficiencia de succínico semialdehído deshidrogenasa, se estudiaron las mutaciones del gen ALDH5A1, en el paciente, encontrándose 2 mutaciones conocidas en heterocigosis: p.Trp204Ter (c.612G>A) y p.Gly268Glu (c.803G>A). Los progenitores resultaron portadores. Se ofreció tratamiento con vigabatrina que los padres rechazaron.
 Corte axial T2; B) Corte coronal T2; C) Corte axial FLAIR a los 2 meses de A y B, y D) Corte coronal T2 (2 meses después de A y B).")
A los pocos meses del diagnóstico presenta cuadro de aparición brusca de debilidad en extremidades de hemicuerpo izquierdo, con predominio en miembro superior con impotencia funcional, que evoluciona a los 4 días a hemiparesia distónica. Se encontraba en tratamiento con risperidona y, además, asociaba decaimiento y episodios de desconexión del medio, de segundos de duración, sin movimientos anormales asociados. Se realizó TAC craneal sin cambios significativos respecto a la imagen previa y el EEG con actividad epileptiforme focal sincrónica e independiente en la región temporal de ambos lados, de predominio derecho. Se retiró tratamiento con risperidona, sin mejoría de la clínica y empeoramiento del comportamiento, se inició con lamotrigina. Posteriormente, en nueva RM, se objetivó evolución de las lesiones previas en globos pálidos sin otros hallazgos (fig. 1). Presentó mejoría de la distonía, persistiendo moderada afectación distal y desaparición de los episodios de desconexión con el tratamiento antiepiléptico.
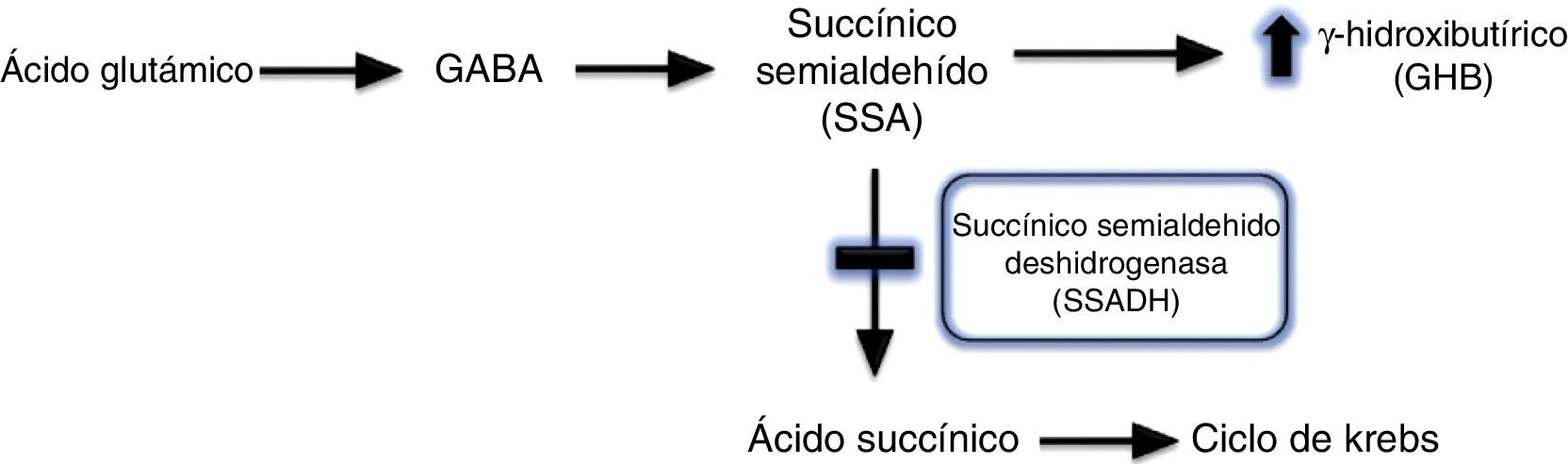
La aciduria gammahidroxibutírica fue descrita por primera vez en 19831, y consiste en el déficit del enzima succínico semialdehído deshidrogenada (SSADH). El GABA mediante un proceso de transaminación se convierte en ácido succínico semialdehído, que a su vez se degrada en ácido succínico por acción de esta enzima, SSDAH. La deficiencia de SSDAH condiciona un aumento del potencial agente neurotóxico 4-hidroxibutírico (GHB) en fluidos fisiológicos1,2 (fig. 2). Es una enfermedad metabólica, de herencia autosómica recesiva, que puede ser originada por diferentes mutaciones del gen ALDH5A1 localizado en el brazo corto del cromosoma 63. Se desconoce la verdadera prevalencia de esta enfermedad, hasta el momento han sido descritos 350 casos en la literatura, la mayoría de edad pediátrica.

La literatura muestra una expresión clínica variable e inespecífica, con manifestaciones neurológicas como hipotonía, hiporreflexia, ataxia no progresiva y retraso psicomotor, de predominio en el área del lenguaje4. Hasta el 50% de estos pacientes presentan convulsiones a lo largo de su evolución, que suelen ser refractarias a tratamiento médico2. Las alteraciones psiquiátricas se describen en la mayoría de los casos, siendo las más limitantes, y abarcan desde rasgos autistas a alucinaciones, agresividad, hiperactividad y trastornos del sueño. La media de edad de diagnóstico son los 6,6 años (rango: 0-25 años)5.
Los estudios electrofisiológicos pueden ser normales en casi la mitad de los pacientes, y las alteraciones que se encuentran en las series de casos publicadas suelen ser actividad epileptiforme focal y generalizada, y enlentecimiento de la actividad de base como hallazgos más frecuentes2. La modalidad de neuroimagen de elección para el estudio de esta enfermedad es la RM, en la que se visualiza de forma característica, hiperintensidad bilateral simétrica en núcleos pálidos en T2. Otros hallazgos estructurales incluyen atrofia cerebro-cerebelar de predominio vermiano, e hiperintensidades corticales, cerebelares y en núcleos dentados2,4,6. Los estudios de espectroscopia muestran un aumento de los niveles de GABA y GHB en sustancia blanca y gris7.
Para establecer el diagnóstico de deficiencia de SSADH, se analiza inicialmente la presencia de GHB o sus isómeros en orina, plasma o líquido cefalorraquídeo mediante espectrometría de masas o cromatografía de gases8. La confirmación se realiza mediante la cuantificación de la actividad del enzima SSADH en leucocitos9 o el estudio genético molecular del gen ALDH5A13. En las familias en las que se detecta la enfermedad es posible el diagnóstico prenatal mediante análisis de ADN o determinación de GHB10 en líquido amniótico o en biopsia de vellosidades coriónicas. Algunos autores11 han propuesto que la detección de GHB en muestras de sangre seca en el periodo neonatal podría ayudar al diagnóstico precoz de la enfermedad.
Las posibilidades de tratamiento son limitadas, aunque se han probado múltiples alternativas, ninguna ha demostrado eficacia en todos los casos. La vigabatrina es un inhibidor de la GABA-transaminasa, y a dosis bajas (25mg/kg/día) disminuye los niveles de GHB en líquido cefalorraquídeo con efectos variables. Autores como Howells et al.12 creen que la inconsistencia de los resultados puede ser debida a que no exista dicha inhibición a nivel periférico. Sin embargo, este tratamiento no está exento de efectos secundarios, siendo el más importante la disminución del campo visual, en ocasiones de forma irreversible. Existen estudios con otros fármacos, pero con resultados aún poco esperanzadores, entre los que se encuentran la taurina, antagonista receptor GHB, NCS-382 y la dieta cetogénica, entre otros13.
Además, deberá realizarse el tratamiento dirigido de los síntomas concomitantes, entre los que se incluyen el de las convulsiones, principalmente con lamotrigina y carbamacepina14, y de las alteraciones del comportamiento con metilfenidato, risperidona y benzodiacepinas15.
El manejo de estos pacientes debe incluir un abordaje multidisciplinar incluyendo medidas no farmacológicas tales como rehabilitación, logopedia, y terapia psicológica y ocupacional.
Aunque la deficiencia de SSADH es una enfermedad rara, debe ser tenida en cuenta en el diagnóstico diferencial de pacientes con retraso psicomotor, alteraciones en el desarrollo del lenguaje, alteraciones del movimiento como ataxia, hipotonía y trastornos de la conducta. En la evolución de la enfermedad, puede aparecer epilepsia, no siendo frecuente la aparición de hemiparesia distónica como la que presentó nuestro paciente.
FinanciaciónNo poseemos financiación.
Conflicto de interesesNo declaramos tener ningún potencial conflicto de intereses.

