



Alzheimer disease (AD) is a major neurodegenerative disorder which eventually results in total intellectual disability. The high global prevalence and the socioeconomic burden associated with the disease pose major challenges for public health in the 21st century. In this review we focus on both existing treatments and the therapies being developed, which principally target the β-amyloid protein.
DiscussionThe amyloidogenic hypothesis proposes that β-amyloid plays a key role in AD. Several pharmacological approaches aim to reduce the formation of β-amyloid peptides by inhibiting the β-secretase and γ-secretase enzymes. In addition, both passive and active immunotherapies have been developed for the purpose of inhibiting β-amyloid peptide aggregation.
ConclusionsProgress in identifying the molecular basis of AD may provide better models for understanding the causes of this neurodegenerative disease. The lack of efficacy of solanezumab (a humanised monoclonal antibody that promotes β-amyloid clearance in the brain), demonstrated by 2 recent Phase III clinical trials in patients with mild AD, suggests that the amyloidogenic hypothesis needs to be revised.
La enfermedad de Alzheimer (EA) es el principal trastorno neurodegenerativo que provoca una discapacidad intelectual total en los pacientes que la presentan. La elevada prevalencia a nivel mundial, así como la elevada carga socioeconómica que conlleva la EA para la sociedad en general, hace que sea considerada un importante problema de salud pública en este siglo xxi. En este trabajo se revisan los tratamientos actuales y en fase de desarrollo que actúan principalmente sobre la proteína β-amiloide.
DiscusiónLa hipótesis amiloidogénica propone que el péptido β-amiloide tiene un papel clave en esta enfermedad. Se han desarrollado varias estrategias farmacológicas diferentes con el objetivo de inhibir la formación de los péptidos β-amiloides, como son los inhibidores de β-secretasa y γ-secretasa. Además, se han desarrollado los tratamientos antiamiloide, que incluyen inmunoterapias pasivas y activas enfocadas a inhibir la agregación del péptido β-amiloide.
ConclusionesLos avances en la identificación de las bases moleculares de la EA pueden servir como modelo para comprender las causas de esta enfermedad neurodegenerativa. Sin embargo, los ensayos clínicos más recientes en 2 ensayos de fase iii con solanezumab, un anticuerpo monoclonal humanizado que promueve el aclaramiento del β-amiloide en el cerebro, indican que este anticuerpo no muestra eficacia en pacientes con EA leve, sugiriendo que hay que replantearse esta hipótesis amiloidogénica de la EA.
Alzheimer disease (AD) is a neurodegenerative disease with a progressive course and the most frequent cause of dementia in the worldwide population older than 65 (50-70% of all dementia cases).1 This chronic and progressive disease gives rise to deficits in multiple brain functions (mainly at the cortical and hippocampal levels); these include memory, thinking, orientation, comprehension, calculation, learning capacity, language, and judgement.2 Alterations contributing to cognitive deficit are accompanied by deterioration in emotional control and social behaviour.
Considering the disease's high prevalence and heavy socioeconomic costs for society at large, AD is regarded as an important public health problem. In fact, statistics indicate that it may constitute the ‘pandemic of the 21st century’, making it a priority for medical research today. Despite the major scientific and clinical advances made in AD research in the last 30 years, the treatments that are currently available are all symptomatic, that is, they lessen the symptoms of the disease by acting on different levels of the neuropathological process.2 Although they can improve patients’ quality of life, none of them is truly able to slow the rapid, fatal progression of the disease.
At present, only 4 currently available drugs have been approved for treating AD. They belong to 2 groups: the acetylcholinesterase inhibitors (AChEI) and the N-methyl-D-aspartate receptors (NMDAR). Donepezil, rivastigmine, and galantamine belong to the AChEI group.2–4 The action mechanism of AChEI drugs is to increase cholinergic transmission by means of inhibiting acetylcholinesterase in the synaptic cleft; this process could increase the cognitive capacity of AD patients somewhat. Memantine is an NMDAR antagonist; it reduces excitotoxicity by blocking that ionotropic receptor, since levels of the excitatory neurotransmitter glutamate are pathologically high in AD. Both drug groups are indicated as treatment for patients in moderate stages of AD.3,4 Nevertheless, it has been shown that none of these approved drugs has a real curative effect; they are only a palliative measure, and their effectiveness decreases over time.
Researchers continue to explore new treatments and therapeutic strategies in order to slow the course of the disease. Above all, these measures are designed for multiple targets and intended for use in the early stages of AD, given the neuropathological complexity of the disease.
If these future treatments are to be effective, doctors must develop new diagnostic techniques that will enable AD diagnosis in its preclinical phase (before symptoms appear) or even able to predict AD before it develops.
AD prevention poses a feasible challenge, but for this goal to be achieved, we must gain a better understanding of the aetiology of AD and how environmental and lifestyle factors affect risk of developing the disease.
Aetiology: proposed hypotheses, risk factors, and protective factorsThe cause or causes of AD are unknown, although different hypotheses have been proposed to explain the complex neurodegenerative process underlying this disease.5–8 Most experts agree that it develops as a result of the presence of multiple risk factors, both modifiable and non-modifiable (age, sex, family history, genetics, environment, and lifestyle), rather than due to a single cause.9–11
At present, the aetiological hypotheses with the most support among the scientific community are the amyloid cascade hypothesis and the phosphorylated tau protein hypothesis.
- –
The amyloid cascade hypothesis6–9 puts forth that the neurodegenerative process observed in brains affected by AD arises primarily from cytotoxic events triggered by the formation, aggregation, and deposition of amyloid beta (Aβ). This hypothesis has received ample support from researchers based on genetic findings from molecular biology studies. These studies have opened new lines of research in the search for AD treatments, including β- and γ-secretase inhibitors and enhancers of α-secretase expression.4
- –
According to this hypothesis, the onset of AD takes place as follows: the amyloid precursor protein (APP) is metabolised by the amyloidogenic pathway, thereby provoking excess production of the Aβ peptide and/or defective elimination of the same.4,5 The Aβ protein is obtained through catabolism of APP, a plasma membrane protein with a single domain (an intracellular and an extracellular region) found in different types of cells. These include neurons, astrocytes, oligodendrocytes, and glial cells.7,8 It is coded for by a gene located on chromosome 21 which can be expressed as any of 8 isoforms; the APP695 isoform is the most abundant in the brain. This protein is cleaved by the enzymes α-, β-, and γ-secretase, plus a protein complex containing the presenilin gene (PSEN1).7 Under physiological conditions and following the non-amyloidogenic pathway, APP is catabolised by α-secretase. This results in the fragment (s)APPα, which remains in the extracellular space, and a carboxy-terminal fragment of 83 amino acids (C83) that remains attached to the plasma membrane. (s)APPα regulates neural excitability, improves synaptic plasticity, learning, and memory, and increases neural resistance to oxidative and metabolic stress.5–8 Nevertheless, in a neuropathological situation, APP is metabolised by the amyloidogenic pathway in which BACE (β-secretase 1) cleaves APP at the N-terminus. In turn, γ-secretase cleaves it at the C-terminus to yield (s)APPβ and Aβ40/42 fragments (which remain in the extracellular space) and a C-terminal fragment with 99 amino acids (C99) that can be transported to the cell interior and translocated to the nucleus. Here, it may induce expression of genes that promote neuronal death by apoptosis.6,7
APP regulates neuronal survival, protection against toxic external stimuli, synaptic plasticity, and cellular adhesion. When it is transformed into Aβ40/42, however, it affects synapse function, decreases neuronal plasticity, alters energy and glucose metabolism, induces oxidative stress and mitochondrial dysfunction, and disturbs cellular calcium homeostasis.7
- –
Differential cleavage by γ-secretase produces different Aβ peptides: Aβ40 is the predominant species, whereas Aβ42 is the main component of senile plaques. Peptide Aβ42 is both more likely to form aggregates and more neurotoxic than peptide Aβ40, which gives rise to the hypothesis that it represents the pathogenic species of Aβ. First, Aβ42 oligomerises and is deposited as senile plaques in the limbic system and associative cortex, thereby exerting toxic effects on neuronal synapses. The second stage involves glial response and activation of astrocytes and surrounding microglia; this releases cytokines or components of the complement system, resulting in inflammatory responses. Furthermore, the neuron experiences oxidative stress; calcium ion homeostasis is disrupted, which leads to hyperactivation of kinase proteins and inactivation of phosphatases. For this reason, tau protein is hyperphosphorylated and forms the neurofibrillary tangles that accumulate in synapses and in neuronal bodies. These structures cause neuronal death by apoptosis and deficit of neurotransmitters. The entire cascade of processes concludes with the onset of dementia.
This being the case, both tau and Aβ proteins (mainly Aβ42) are recognised as the main targets for disease-modifying therapy in AD.4 In line with this hypothesis, AD might be prevented or treated effectively by decreasing Aβ42 production and tau protein phosphorylation; by preventing the deposition or misfolding of these proteins; by neutralising or removing deposits or toxic misfolded forms of these proteins; or a combination of the above strategies.4–9
At present, researchers are proposing alternative hypotheses that range from altered mitochondrial activity to neuroinflammation or the role played by metabolism, specifically cholesterol and insulin.10–14 The latest is the dendritic hypothesis of AD.15 This array of theories testifies to the complexity of the disease, which is further complicated by the fact that the mechanism underlying neuronal apoptosis is not fully understood.
Therapeutic strategies used in developing disease-modifying treatments for ADSince evidence suggests that the number of cases of AD will continue to rise in the next few decades, doctors need to develop a treatment able to modify the course of the disease more effectively.
In the period between 1998 and 2011, some 100 compounds tested to determine whether they could modify the course of AD failed to demonstrate efficacy while in the clinical development phase.1,3 The reason why these compounds failed to show efficacy could be the complexity of the disease, which is known for its multifactorial aetiology and intricate pathophysiology. Finding a suitable drug that is also effective in the entire trial population is no easy task.
Although several key aspects of AD pathogenesis have yet to be resolved, scientific progress in the last 25 years has allowed us to establish different rational strategies for developing treatments that may be able to modify the course of AD. Therefore, out of all of the treatment strategies being developed, those aimed at reducing the formation of Aβ42 and the phosphorylation of tau protein are the most important.3 The greatest advances in this field have had to do with these 2 types of processes, and findings in this area may be key to AD treatment in the near future.
Anti-amyloid strategiesOver the past 2 decades, research has focused mainly on the role of Aβ in line with the amyloidogenic hypothesis. Scientists have dedicated their best efforts to the challenge of developing effective drug treatments for AD.4,6 However, the repeated clinical failures of the compounds being developed have led researchers to question that hypothesis. At present, both new compounds and new tools for AD diagnosis are being investigated. It is believed that past failures may be due to the lack of biomarkers able to facilitate patient recruitment for clinical trials before those patients have reached very advanced stages of disease in which any type of therapeutic intervention would be fruitless.1
Different anti-amyloid strategies are intended to act on different steps in APP metabolism.
Decrease in Aβ peptide production: secretase inhibitorsResearch efforts have focused on modulating enzymatic pathways responsible for abnormal APP processing in an attempt to decrease production of Aβ. In other words, they target the inhibition of γ- and/or β-secretase and the activation of α-secretase.
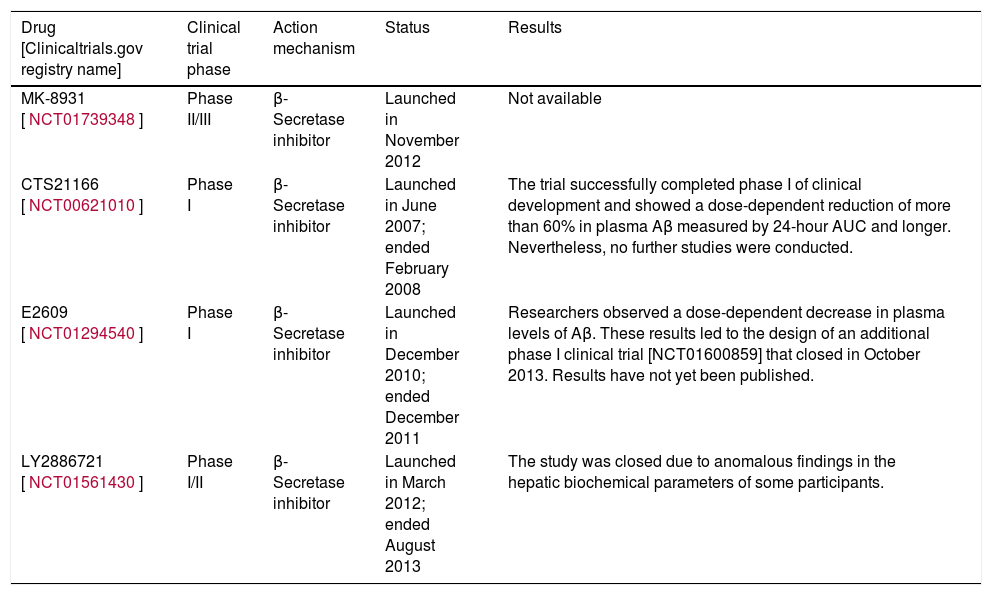
Inhibitors of β-secretase (BACE1)The β-secretase enzyme is responsible for initiating the amyloidogenic pathway for processing APP.7 Developing inhibitors of this enzyme is a major challenge because, in addition to APP, β-secretase has many more substrates. These include neuregulin-1, which is involved in myelination of the peripheral nerves.16–18 For this reason, non-specific inhibition of the enzyme may result in adverse effects.16 On the other hand, the largest problem has to do with the enzyme's structure. Since it belongs to the aspartic protease class, the inhibitor must be a large, hydrophilic molecule; as such, it will not be able to cross the blood-brain barrier easily.19 At present, scientists are examining various compounds with the aim of overcoming these obstacles and obtaining a drug that will be effective against AD.19 Recent studies indicate that 2 inhibitors of β-secretase, E2609 and MK-8931, are extremely effective at reducing Aβ levels (by 80%-90%) in human CSF.17–19
Table 1 shows some of the compounds currently in the clinical development phase.
Clinical trials performed with β-secretase inhibitors.
| Drug [Clinicaltrials.gov registry name] | Clinical trial phase | Action mechanism | Status | Results |
|---|---|---|---|---|
| MK-8931 [NCT01739348] | Phase II/III | β-Secretase inhibitor | Launched in November 2012 | Not available |
| CTS21166 [NCT00621010] | Phase I | β-Secretase inhibitor | Launched in June 2007; ended February 2008 | The trial successfully completed phase I of clinical development and showed a dose-dependent reduction of more than 60% in plasma Aβ measured by 24-hour AUC and longer. Nevertheless, no further studies were conducted. |
| E2609 [NCT01294540] | Phase I | β-Secretase inhibitor | Launched in December 2010; ended December 2011 | Researchers observed a dose-dependent decrease in plasma levels of Aβ. These results led to the design of an additional phase I clinical trial [NCT01600859] that closed in October 2013. Results have not yet been published. |
| LY2886721 [NCT01561430] | Phase I/II | β-Secretase inhibitor | Launched in March 2012; ended August 2013 | The study was closed due to anomalous findings in the hepatic biochemical parameters of some participants. |
The enzyme γ-secretase is responsible for the final stage in APP processing by the amyloidogenic pathway; it produces peptides Aβ40 and Aβ42. Although achieving inhibition of this enzyme in 2001 was seen as a breakthrough with promising implications for disease modification, and this was the first time production of Aβ had been decreased in vivo, developing inhibitors of γ-secretase and inhibitors of β-secretase will entail the same challenges.19–21
The enzyme γ-secretase acts to process other proteins in addition to APP, including Notch protein, which regulates cell proliferation, development, differentiation, and communication, as well as cell survival.20,21 For this reason, nonspecific inhibition of this enzyme will result in severe adverse effects that seriously limit clinical trials.
One of the drugs that has been developed is semagacestat (LY450139), an inhibitor of functional γ-secretase that was shown to lower levels of Aβ in blood and CSF in human studies.22 The clinical trial measured Aβ plaques in the brain by means of a scanner able to create images of amyloid plaques. Results from this study and other similar studies (NCT00762411; NCT01035138; NCT00762411) showed that semagacestat did not halt the slow progression of the disease; furthermore, use of this drug was associated with poorer cognition and decreased ability to carry out activities of daily living.22 Another drug that has been tested is avagacestat (NCT00810147; NCT00890890; NCT00810147; NCT01079819); multiple clinical trials have evaluated its pharmacokinetics and efficacy against AD.23–25
One solution devised to prevent the adverse effects derived from these inhibitors of the γ-secretase enzyme was the use of selective γ-secretase modulators (GSMs), which block the enzyme to alter APP processing while sparing signalling on other pathways, such as the Notch pathway.21 The development of selective GSMs began with the observation that different non-steroidal anti-inflammatory drugs (NSAIDs) decreased levels of the Aβ42 peptide in cell cultures and in rats.26,27 These drugs include ibuprofen, sulindac, indometacin, and flurbiprofen. R-flurbiprofen (tarenflurbil) inhibits cyclooxygenase-1 to a much lesser extent, and it is now in a phase III clinical trial as a potential treatment for AD. In contrast, tarenflurbil and ibuprofen both failed to show efficacy in their respective clinical trials.27,28
CHF5074 is a nonsteroidal anti-inflammatory derivative that does not act as an inhibitor of cyclooxygenase.29 In vitro studies show that CHF5074 acts as a modulator of γ-secretase, preferably by inhibiting production of Aβ42.30,31 As previously stated, long-term use of NSAIDs offers some protection against AD, and this observation led to studies of the effect of NSAIDs on the production of Aβ42. Nevertheless, the negative results delivered by clinical trials of NSAIDs indicate that protection against AD is not a general benefit offered by all drugs of this type.
One example of a selective GSM is NIC5-15, a naturally occurring molecule. Also known by the name of pinitol, NIC5-15 is a natural cyclic sugar alcohol.32 It is found in soy and in several other plants and fruits. Furthermore, pinitol acts as an insulin sensitiser. This compound modulates γ-secretase by reducing Aβ production, but it does not affect cleavage of the Notch-γ-secretase substrate.32,33 The compound has been found to decrease functional deficit and improve cognitive memory in preclinical models of AD neuropathology.33 Animal models and drug trials in humans have both shown that NIC5-15 is safe and acts as an insulin sensitiser.32 In preclinical studies testing doses higher than those previously administered in clinical trials, researchers observed that NIC5-15 interfered with Aβ deposition. This finding indicates that NIC5-15 may be an appropriate agent for treating AD for 2 reasons: it is a Notch-sparing γ-secretase inhibitor, and it may also be an insulin sensitiser. Researchers are now examining its potential role as an inhibitor of the inflammatory process, especially referring to inhibition of microglial activation.
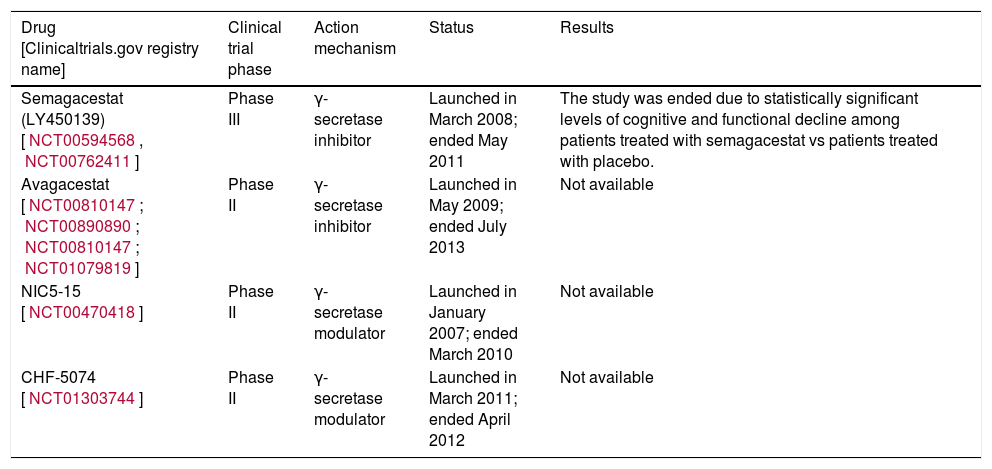
Different companies are running studies on inhibitors and modulators of γ-secretase; some are listed in Table 2.
Clinical trials performed with γ-secretase inhibitors.
| Drug [Clinicaltrials.gov registry name] | Clinical trial phase | Action mechanism | Status | Results |
|---|---|---|---|---|
| Semagacestat (LY450139) [NCT00594568, NCT00762411] | Phase III | γ-secretase inhibitor | Launched in March 2008; ended May 2011 | The study was ended due to statistically significant levels of cognitive and functional decline among patients treated with semagacestat vs patients treated with placebo. |
| Avagacestat [NCT00810147; NCT00890890; NCT00810147; NCT01079819] | Phase II | γ-secretase inhibitor | Launched in May 2009; ended July 2013 | Not available |
| NIC5-15 [NCT00470418] | Phase II | γ-secretase modulator | Launched in January 2007; ended March 2010 | Not available |
| CHF-5074 [NCT01303744] | Phase II | γ-secretase modulator | Launched in March 2011; ended April 2012 | Not available |
Activation of the α-secretase enzyme leads to APP being processed by the non-amyloidogenic pathway, which thus decreases the amount of APP available to the amyloidogenic pathway. The result is a soluble Aβ peptide that has been shown to play a role as a neuroprotector and stimulator of synaptogenesis.
As such, α-secretase activation provides an attractive strategy for the development of disease-modifying drugs. Different compounds potentially able to stimulate the non-amyloidogenic pathway have been investigated. These include acetylcholine muscarinic receptor, glutamatergic, and serotonergic agonists, and protein kinase C activators. Nevertheless, scientists have not found any large compounds able to effectively modulate this pathway in animal models, and therefore very few such compounds have reached the clinical trial stage.
Epigallocatechin gallate (EGCG), a polyphenolic flavonoid extracted from green tea leaves, is considered to be the key bioactive ingredient in green tea. It is reported to have beneficial clinical effects ranging from anti-neoplastic to anti-inflammatory and neuroprotective properties; additionally, it may have a beneficial effect on cognitive function.34 EGCG is believed to inhibit formation of toxic misfolded oligomers of Aβ, in addition to activating α-secretase. A clinical trial (NCT00951834) is currently underway to evaluate the efficacy of EGCG in early stages of AD.
Briostatin 1 is a PKC modulator that also appears to exert an immunomodulatory effect.35 It has been shown to increase cognitive ability in laboratory animals.35
Etazolate (EHT 0202) stimulates the neurotrophic action of α-secretase; it also inhibits neuronal death induced by Aβ which alleviates symptoms and modifies disease progression. A recent phase II clinical study in 159 patients with mild to moderate AD showed EHT 0202 to be safe and well tolerated overall.36 These encouraging initial results provide further support for developing EHT 0202 to evaluate its clinical efficacy and confirm its tolerability in a large cohort of patients with AD, and over a longer period of time.36
Acitretin is a retinoid that acts as a retinoic acid receptor agonist; it is mainly used to treat severe psoriasis.37 In preclinical models it increases expression of ADAM-10, the α-secretase of human APP.37–39 Acitretin has been observed to activate the non-amyloidogenic pathway of APP processing in neuroblastoma cells, and it reduces levels of Aβ in transgenic APP/PSI mice.37–39
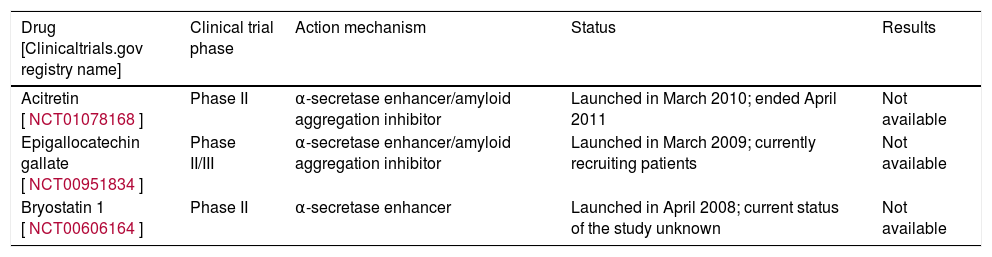
Table 3 lists 2 of these compounds that have reached the clinical development stage: EGCG and briostatin-1.
Clinical trials performed with α-secretase inhibitors.
| Drug [Clinicaltrials.gov registry name] | Clinical trial phase | Action mechanism | Status | Results |
|---|---|---|---|---|
| Acitretin [NCT01078168] | Phase II | α-secretase enhancer/amyloid aggregation inhibitor | Launched in March 2010; ended April 2011 | Not available |
| Epigallocatechin gallate [NCT00951834] | Phase II/III | α-secretase enhancer/amyloid aggregation inhibitor | Launched in March 2009; currently recruiting patients | Not available |
| Bryostatin 1 [NCT00606164] | Phase II | α-secretase enhancer | Launched in April 2008; current status of the study unknown | Not available |
The vast body of evidence pointing to the neurotoxic and synaptotoxic activity of amyloid deposits provides the scientific basis for the development of Aβ peptide aggregation inhibitors.
The sole Aβ aggregation inhibitor to have reached a phase II trial is a synthetic glucosaminoglycan, 3-amino-1-propanesulfonic acid (3-APS, Alzhemed, tramiprosate).40,41 This drug was designed as an inhibitor or antagonist of the interaction between Aβ and endogenous glucosaminoglycans. Glucosaminoglycans have been shown to promote Aβ aggregation by contributing to the formation of amyloid fibrils and the establishment of plaque-type deposits.41 Nevertheless, disappointing results from a phase III trial in 2007 led to the suspension of the European phase III trial.
Colostrinin, a polypeptide complex rich in proline and derived from ovine colostrum, inhibits both Aβ aggregation and Aβ toxicity in cell cultures; it also improves cognitive performance in animal models.42 Although a phase II trial indicated slight improvements on the Mini–Mental State Examination in patients with mild AD who were treated for 15 months, this beneficial effect was not observed in the following 15-month period of continued treatment.43
The compound known as scyllo-inositol is able to stablise oligomeric aggregates of Aβ and inhibit Aβ toxicity in the murine hippocampus. An 18-month clinical trial has been carried out to determine the dosage, safety, and efficacy of scyllo-inositol (ELND005) in patients with mild to moderate AD. This trial evaluated 3 doses of ELND005 (250 mg, 1000mg, and 2000mg) to determine that 250mg was the most appropriate. Further long-term clinical studies in patients with AD will be needed in order to obtain evidence sufficient to demonstrate or rule out any beneficial effects of ELND005 as treatment for this disease.44
Multiple compounds with antiaggregant effects are now being evaluated, including PBT1 (clioquinol) and PBT2. Clioquinol was examined as a potential AD treatment, since this compound inhibits the interaction between metals and the Aβ peptide in the brain.45 It has been suggested that the increase in bioactive metals occurring in the ageing brain could accelerate the formation of amyloid plaques, as well as any neurotoxic oxidative processes. The basic reason for holding trials of clioquinol was that it was thought to prevent Aβ deposits and also restore cellular homeostasis of such ions as copper and zinc. Nevertheless, these compounds did not show enough efficacy in phases II and III of the clinical development stage.
Compounds promoting elimination of amyloid aggregates and depositsThe third strategy using the amyloidogenic pathway targets clearance of amyloid aggregates and deposits. Three different approaches have been used to this end.
Activation of enzymes responsible for the breakdown of amyloid plaqueAmyloid aggregates and plaques are broken down by different proteases, including neprilysin, insulin-degrading enzyme, plasmin, endothelin converting enzyme, angiotensin converting enzyme, and metalloproteinase 9.46,47 The level of these enzymes drops in AD, and this situation contributes to amyloid plaque formation and accumulation.46 Although this seems to provide an attractive amyloid-fighting strategy for use in the development of disease-modifying drugs, no protease activators have been evaluated at present because these compounds lack specificity.
Modulation of amyloid β transport from the brain to the peripheral circulationTransport of Aβ between the central nervous system (CNS) and the peripheral circulation is regulated by the following elements: 1) apolipoproteins, including APOE¿4 that promotes the transfer of Aβ from the bloodstream to the brain; 2) low density lipoprotein receptor-related proteins (LRP) that increase transfer of Aβ from the brain to the bloodstream; and 3) the receptor for advanced glycation end products (RAGE), which facilitates penetration of Aβ in the CNS.48–51
Although there are different proposed strategies for increasing Aβ transport from the brain to the peripheral circulation, such as peripheral administration of LRP, only the compounds intended to inhibit or modulate RAGE have reached the clinical development stage. These end products include PF-04494700,52 which failed a phase II clinical trial, and TTP4000, currently undergoing a phase I clinical trial (NCT01548430). The study ended in February 2013 and results have not yet been published.
Specific anti-amyloid immunotherapyActive immunotherapy: immunotherapy comprises the third strategy intended to improve Aβ clearance, and the most studied method of reducing the amyloid burden in AD. Active immunisation (vaccination), whether with Aβ42 (the predominant form of Aβ in the amyloid plaques forming in AD) or with other synthetic fragments, has shown success in transgenic mouse models of AD. These trials are typically based on stimulating T and B cells and generating an immune response by activating the phagocytic capacity of microglia. While initial results from these trials were promising, the studies have been partially suspended since some of the patients developed meningoencephalitis.
When the first immunisation consisting of the Aβ peptide with 42 amino acids was tested in patients, researchers observed that it elicited neuroinflammatory processes, such as aseptic meningoencephalitis, as the result of an anti-AN1792 autoimmune response mediated by T cells.53 These adverse effects are the reason why the phase II clinical trials were halted.
By designing second generation vaccines, researchers attempted to prevent a nonspecific immune response to immunisation with complete Aβ peptides (Aβ1-42). These vaccines employed shorter segments of the Aβ peptide (Aβ1-6) that would promote a humoural response to the cellular immune response.
CAD 106, designed by Novartis, was the first second-generation vaccine to reach the clinical development stages.54 This drug recently completed phase II clinical trials in which researchers observed a specific response by Aβ in 75% of the treated patients without any adverse inflammatory responses. ACC-001 recently completed phase II trials (NCT01284387 and NCT00479557) and another phase II trial is underway (NCT01227564); nevertheless, the pharmaceutical company has decided to discontinue this line of research. Additional immunisations, such as ACI-24 (an Aβ1-15 tetra-palmitoylated peptide reconstituted in a liposome), MER5101, and AF205 are currently in preclinical stages of development since they are still undergoing laboratory testing.55–57
Passive immunisation: another type of immunotherapy under study involves passive administration of monoclonal or polyclonal antibodies targeting Aβ. The technique consists of intravenous administration of Aβ antibodies. This strategy provokes an anti-Aβ immune response with no need for a proinflammatory reaction mediated by T-cells.57 Studies in transgenic animals have shown that passive immunisation reduces not only the amyloid burden in neurons, but also cognitive deficit, even before neuronal amyloid plaques have been eliminated.58 These findings may be attributed to neutralisation of soluble amyloid oligomers, which are increasingly believed to play a fundamental role in the pathophysiological cascade occurring in AD.57,58
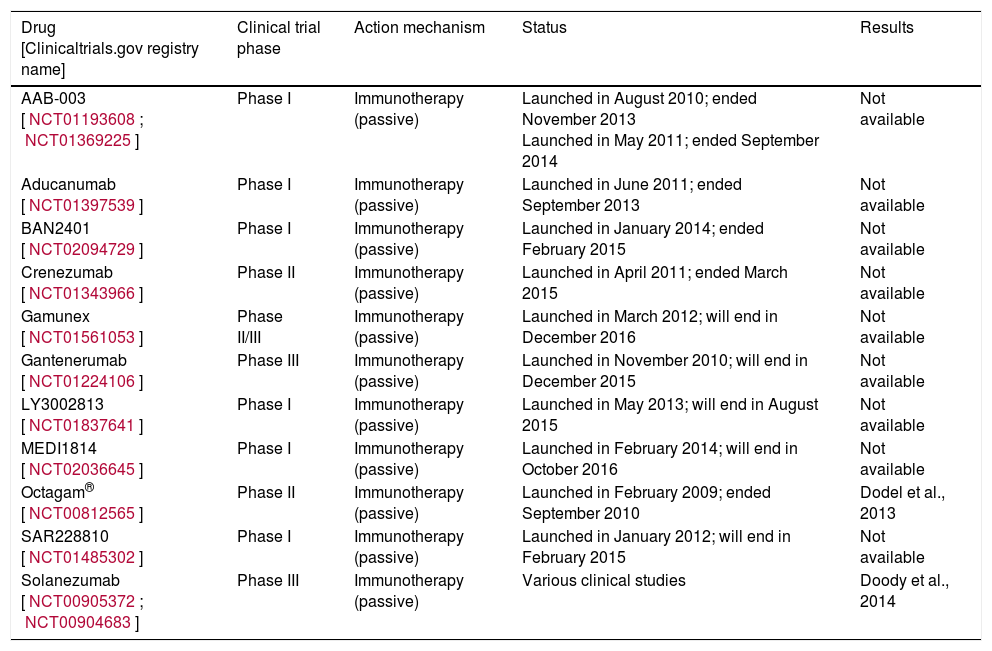
Bapineuzumab and solanezumab, the 2 monoclonal antibodies to have reached the most advanced stages of clinical development, both failed to show the desired clinical benefits in patients with mild to moderate AD in 2 phase III clinical trials in 2012. Bapineuzumab is a humanised monoclonal antibody against the N-terminus of the Aβ protein (Aβ1-5); solanezumab is a humanised monoclonal antibody designed to bind the central part of Aβ protein (Aβ12-28).59,60 Although bapineuzumab reduced key AD biomarkers including cerebral amyloid plaque and CSF phosphorylated tau protein, it failed to demonstrate significant cognitive improvement in 2 clinical trials.57,61Table 4 lists the clinical trials that have been completed.
Clinical trials performed with Aβ aggregation inhibitors.
| Drug [Clinicaltrials.gov registry name] | Clinical trial phase | Action mechanism | Status | Results |
|---|---|---|---|---|
| AAB-003 [NCT01193608; NCT01369225] | Phase I | Immunotherapy (passive) | Launched in August 2010; ended November 2013 Launched in May 2011; ended September 2014 | Not available |
| Aducanumab [NCT01397539] | Phase I | Immunotherapy (passive) | Launched in June 2011; ended September 2013 | Not available |
| BAN2401 [NCT02094729] | Phase I | Immunotherapy (passive) | Launched in January 2014; ended February 2015 | Not available |
| Crenezumab [NCT01343966] | Phase II | Immunotherapy (passive) | Launched in April 2011; ended March 2015 | Not available |
| Gamunex [NCT01561053] | Phase II/III | Immunotherapy (passive) | Launched in March 2012; will end in December 2016 | Not available |
| Gantenerumab [NCT01224106] | Phase III | Immunotherapy (passive) | Launched in November 2010; will end in December 2015 | Not available |
| LY3002813 [NCT01837641] | Phase I | Immunotherapy (passive) | Launched in May 2013; will end in August 2015 | Not available |
| MEDI1814 [NCT02036645] | Phase I | Immunotherapy (passive) | Launched in February 2014; will end in October 2016 | Not available |
| Octagam® [NCT00812565] | Phase II | Immunotherapy (passive) | Launched in February 2009; ended September 2010 | Dodel et al., 2013 |
| SAR228810 [NCT01485302] | Phase I | Immunotherapy (passive) | Launched in January 2012; will end in February 2015 | Not available |
| Solanezumab [NCT00905372; NCT00904683] | Phase III | Immunotherapy (passive) | Various clinical studies | Doody et al., 2014 |
At present, new phase III clinical trials are being carried out on solanezumab, both in patients with AD (NCT01127633 and NCT01900665) and in elderly, asymptomatic patients (A4) at high risk of losing memory (NCT02008357). Another monoclonal antibody, gantenerumab, is being tested by the Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU) to evaluate its disease-modifying potential in people at risk of developing early-onset AD due to an autosomal dominant mutation.62,63 More specifically, in phase III of the trial, patients with mild to moderate AD were given infusions of 400mg of solanezumab or placebo once a month over a period of 80 weeks. Results seem to indicate a tendency towards cognitive improvement in the solanezumab group in patients with mild AD, but this tendency does not appear to be statistically significant. For these reasons, we will have to wait until more results are available.
At the same time, several phase III clinical trials are being conducted to test the efficacy and safety of gantenerumab in patients with mild AD (NCT02051608) and AD in its prodromal stages (NCT01224106). Gantenerumab is a fully human IgG1 antibody designed to have a high affinity for binding to a conformational epitope expressed on Aβ fibrils.62,63 The therapeutic basis for this antibody is that it acts by breaking down amyloid plaque through a process involving microglial recruitment and activation of phagocytosis. Experimental studies in transgenic mice support this hypothesis.64
Crenezumab (MABT5102A) is another humanised monoclonal antibody to have reached the clinical development stage.65 April 2014 marked the end of a phase II clinical trial in which the efficacy and safety of this drug was evaluated in patients with mild to moderate AD (NCT01343966), but results are not available. Phase II studies of crenezumab are currently underway; the most recent was launched in 2013 for the purpose of evaluating drug efficacy and safety in asymptomatic carriers of the autosomal dominant mutation of PSEN1 (NCT01998841).
Other monoclonal antibodies against Aβ to have been developed to date include PF-04360365 (ponezumab), which targets the free C-terminus of Aβ, specifically Aβ34-41; MABT5102A which has equal affinity for binding monomers, oligomers, Aβ, and fibrils; and GSK933776A which is similar to bapineuzumab in that it targets the N-terminus of Aβ.65 Other passive immune agents are being developed, such as GSK933776A, NI-101, SAR-228810, and BAN-2401; most are in phase I clinical trials (see Table 4).
Gammagard™ is a human plasma antibody solution. It has demonstrated a safe profile for use against certain autoimmune disorders in humans. Gammagard has also been evaluated as treatment for AD in a small patient sample (NCT00818662). These intravenously-delivered immunoglobulin mixtures contain a small fraction of polyclonal antibodies targeting the Aβ peptide. This is thought to be able to combat the synaptic toxicity caused by Aβ.66–68 Furthermore, this intravenous immunoglobulin exerts an immunomodulatory effect, in addition to favouring phagocytosis by microglia.68
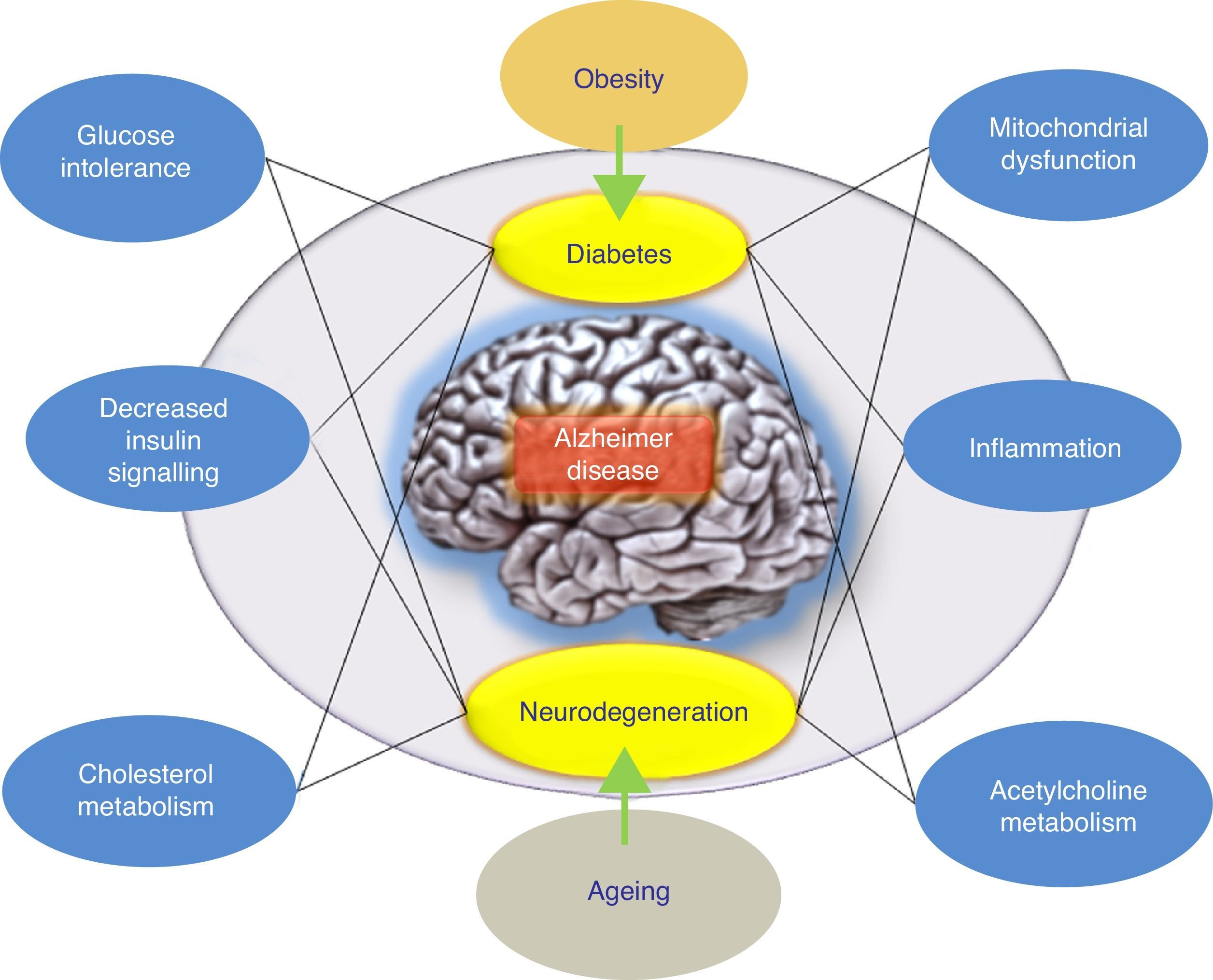
ConclusionsThere have been numerous attempts to treat AD by reducing levels of Aβ in the brain. The overall results at this time have delivered data indicating that anti-amyloid drugs, as a specific drug class, may have a deleterious effect on the symptoms of the disease. On the other hand, results from different completed studies support systematic classification of these treatment approaches according to the underlying mechanism rather than grouping all anti-amyloid treatments together. Furthermore, alternative hypotheses have been proposed to explain the failure of the amyloidogenic hypothesis. These include the adaptive response hypothesis, stating that Aβ may aggregate due to an adaptive response to chronic stress stimuli at the cerebral level.9 To this end, these stress stimuli would constitute the pathogenic trigger signals or pathways for late onset of AD, and in this case, they would be the right candidates for therapeutic interventions. In this scenario, the appropriate drug treatment for slowing AD would act upon these stress stimuli. These stimuli include oxidative stress, metabolic dysregulation (cholesterol homeostasis, insulin resistance, etc.), genetic factors, and inflammatory response (Fig. 1). Each of these stimuli is able to provoke a response by which more Aβ is produced, and the nature of this response is what determines the clinical course of the disease. With this in mind, recent studies are evaluating intranasal insulin, a promising strategy in AD treatment. Favourable results would confirm the hypothesis according to which the Aβ peptide is not the only pathogenic agent in AD.
. According to this hypothesis, treatment would consist of drugs that restore insulin tolerance. The right drug treatment for slowing AD would act on these stress stimuli (the inflammatory response, mitochondrial changes, etc.).")
Model offering a potential explanation for late-onset Alzheimer disease. The adaptive response hypothesis proposes that Aβ can accumulate as an adaptive response to chronic stress stimuli such as metabolic dysregulation (altered cholesterol homeostasis or insulin resistance). According to this hypothesis, treatment would consist of drugs that restore insulin tolerance. The right drug treatment for slowing AD would act on these stress stimuli (the inflammatory response, mitochondrial changes, etc.).
This study was financed by the Generalitat de Catalunya (2009/SGR00853), the Spanish Ministry for Science and Innovation (SAF2011-23631), CIBERNED Instituto de Salud Carlos III, and the PROMETEO programme of the Government of Ecuador.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Folch J, Ettcheto M, Petrov D, Abad S, Pedrós I, Marin M, et al. Una revisión de los avances en la terapéutica de la enfermedad de Alzheimer: estrategia frente a la proteína β-amiloide. Neurología. 2018;33:47–58.
