



Isodicentric chromosome 15 (idic[15]), also known as inverted duplication 15 (inv dup 15) or partial tetrasomy 15q (OMIM 608636), is a clearly-defined clinical entity that presents with central hypotonia, developmental delay, intellectual impairment, epilepsy, autistic behaviour, and certain minor phenotypic abnormalities. Its estimated incidence rate is 1 case per 30000 births and it affects both sexes equally.1 From a cytogenetic viewpoint, the disorder is defined as a mirror-image duplication of the piece of chromosome 15 between the end of the short arm and region 12–13 of the long arm (idic[15][pter→q12–13::q12–13→pter]), including region q11 (q11→q13). This location contains the critical regions for Prader-Willi syndrome (PWS) and Angelman syndrome (AS). In most of the cases studied to date, the extra chromosome is maternal in origin and related to phenotypic abnormalities and advanced maternal age.2–4
Chromosomal region 15q1q13 is known for its instability, which is secondary to the presence of repetitive DNA sequences. It is highly susceptible to the formation of clinically relevant DNA rearrangements, such as the formation of supernumerary markers formed by mirror-image duplication of chromosome 15. This phenomenon may result in tetrasomy of the short arm of chromosome 15 (15p) or partial tetrasomy of the long arm of chromosome 15 (15q).5 Phenotypic variability in these patients tends to be determined by the specific region involved in each chromosomal rearrangement. We report a case of isodicentric chromosome 15, with multiple physical anomalies and epilepsy, which involves 15q11.2 as a breakpoint.
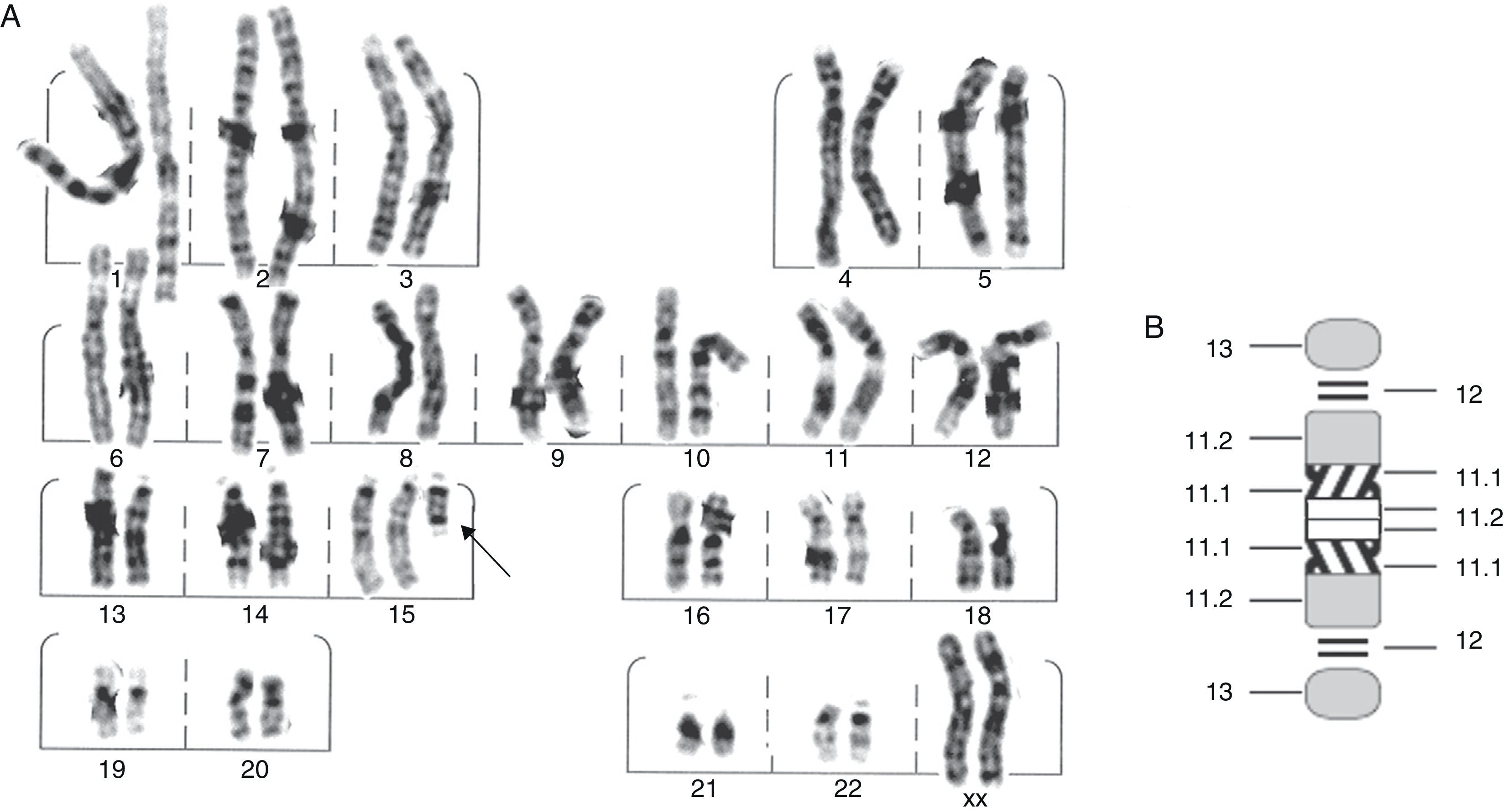
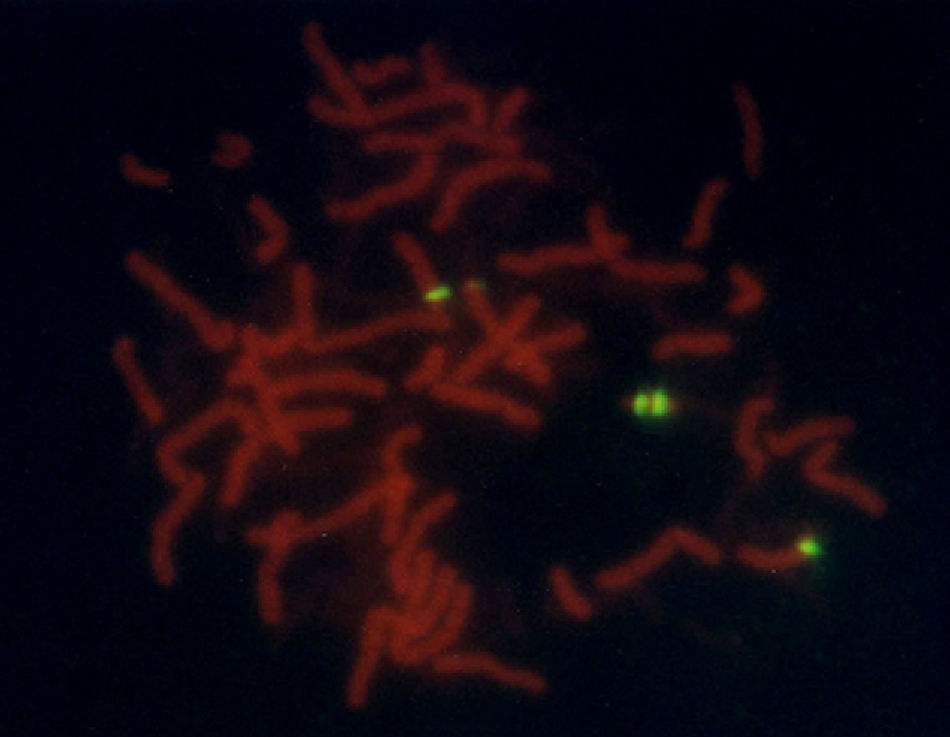
A female patient was referred to the genetics department with a working diagnosis of seizures and minor abnormalities. Physical examination revealed oval-shaped face, broad nose, slightly slanting palpebral fissures, prominent ears, moderate midfacial hypoplasia, mild microagnathia, long limbs, and a tendency toward genu valgum and talipes equinovarus. Biochemical testing showed normal amino acid levels; karyotyping confirmed the presence of a minichromosome that could belong to part of either chromosome 14 or 15. Based on phenotypic evidence, it was reported as partial trisomy 15 (15pter→15q1:) (Fig. 1). To confirm the diagnosis, we performed FISH analysis with a centromeric probe for chromosome 15. In both nuclei and in chromosome 15 copies, we observed 4 fluorescent points, one in each normal copy of chromosome 15 and two in the minichromosome, which indicated the presence of dicentric chromosome 15 (Fig. 2). This was interpreted as partial tetrasomy 15q due to inverted duplication of segment 15pter→15q11.2: 47,XX,+der(15)(pter→q1:).ish invdup(15)(pterq11.2)(D15Z1++) or of 47,XX,+der(15)(pter→q1:).ish invdup(15)(pter→q11.2::q11.2→pter)(D15Z1++).
 G-banding karyotype showing an extra chromosome derived from chromosome 15 (arrow). (B) Ideogram of the extra chromosome, identifying breakpoints.")

In general, patients with isodicentric 15 present developmental delays and begin sitting up between 10 and 20 months and walking between 2 and 3 years. They also display cognitive deficit in the areas of comprehension and expressive language (some exhibit echolalia). Their behaviour is frequently referred to as autistic or autistic-like. Epilepsy characteristics vary, and no specific type predominates except for Lennox-Gastaut syndrome, which Battaglia described in 4 patients in 1997. These patients presented tonic-atonic, tonic–clonic, and absence seizures. Other types of seizures include partial complex seizures, generalised myoclonic seizures with adult onset, generalised tonic–clonic seizures, and benign epilepsy with centrotemporal spikes. Patient age at seizure onset varied from 6 months to 9 years. It has been suggested that the degree of intellectual impairment and psychomotor delay could be correlated with the severity and intractability of convulsions. However, few studies have specifically addressed this subject.2 In general, patients’ facial appearance is normal. Non-specific features may be present, including downward-slanting palpebral fissure, epicanthic fold, deep-set eyes, low-set ears with or without backwards rotation, high palate, broad nose, anteverted nostrils, clinodactyly of the fifth digit of the hand and partial sindactyly of the second and third digits of the feet.3 Our patient did not present any of the neurological changes reported except for convulsions and minor physical anomalies, plus a few more significant features including talipes equinovarus.
Chromosome 15, and more specifically, the 15q11–q13 locus, is a cluster of genes whose genetic imprint is essential for normal neural development in mammals. As mentioned previously, this locus is extremely unstable, especially at the 5 common breakpoints (from BP1 to BP5), which may give rise to genomic rearrangements including deletions and duplications. One of the suggested mechanisms for idic(15) involves U-type recombination between homologous chromosomes, followed by the disjunction and inactivation of one of the centromeres. The most common form of idic(15) is characterised by an asymmetric recombination event between BP4 and BP5. This produces tetrasomy of the region between the centromere and BP4 and trisomy of the region between BP4 and BP5.3 Maternal-origin interstitial duplications and supernumerary isodicentric chromosomes produce various types of neurodevelopmental disorders which sometimes present with autistic traits. This duplication is the main cytogenetic cause of autism, and responsible for 1% to 3% of all cases.6 In the same way, we are aware of the environmental and genetic factors that contribute to the characteristics of each clinical case. This is also true for most behavioural syndromes which have been related to a wide array of infectious diseases, such as embryopathy induced by rubella or encephalitis.7 It should be noted that the second leading cause of isodicentric 15 is intrachromosomal triplication. This type of anomaly may arise in response to a 2-step process taking place during meiosis. First, a U-type exchange occurs (rather than a normal, X-type exchange), resulting in an inverted dicentric duplication which subsequently recombines with a normal copy of chromosome 15 to form the triplication. The anomaly may also form as the result of a U-type exchange between 3 chromatids in a single step.8,9
The large majority of chromosome 15 duplications (dup[15]) and SMC(15) containing the critical region for PWS/AS are maternal in origin. This fact implies that paternal-origin duplication is either uncommon and lethal, meaning that the fetus would rarely be brought to term, or else not capable of causing phenotypic effects, meaning that the duplication would go undetected. However, we cannot rule out the possibility that apparent lack of a phenotype could be secondary to a position effect that would lead to the inactivation of the transcriptionally active gene or genes near the breakpoint in 15q.10,11
Modern basic and molecular cytogenetic techniques have allowed us to establish a correlation between the size of the duplicated fragment and the resulting phenotype. Small duplications in regions that are very close to the centromere produce no effects, whereas duplications affecting more extensive areas, with breakpoints in q12 and q13, give rise to intellectual impairment, autism, or seizures.1 In this particular case, the affected region, 15q11.2, includes genes that may potentially contribute to epilepsy, such as genes encoding gamma-aminobutyric acid (GABA) receptor subunits. GABA is known as the main inhibitory neurotransmitter in mammal brains; however, it also displays an excitatory effect in mature neurons of the hypothalamic suprachiasmatic nucleus. This dual activity may be regulated by diurnal oscillations in intracellular chloride concentrations. Therefore, excessive duplication and expression of these neurotransmitter receptors may contribute to the induction of disease, or the development of hyperactivity and aggressive behaviour, as has been observed in adults with epilepsy and idic(15).5,11,12
Since physical examination of these patients reveals no dysmorphic features or minor anomalies in most cases, chromosome analysis may never be carried out. As a result, the underlying chromosomal anomaly is not diagnosed in many such patients. For that reason, high-resolution karyotyping and molecular cytogenetic analysis are recommended for all patients with developmental delays, epilepsy (especially drug-resistant epilepsy) and autism, whether or not they present dysmorphic features, in order to rule out chromosomal alterations.
This case report was written for academic and pedagogical purposes in light of the low incidence rate of this disease (1:30000 live births) and the importance of the complementary study performed on this patient. That study enabled doctors to assign a final diagnosis and thus offer appropriate genetic counselling to the patients’ parents.
Please cite this article as: Gordillo-González G, et al. Paciente con síndrome convulsivo y tetrasomía parcial del cromosoma 15. Neurología. 2013;28:191–3.
This article was presented in poster format at the 11th Colombian Congress on Human Genetics, Medellín, October 2010.

Neurología (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals