



El síndrome de duplicación invertida del cromosoma 15 (invdup[15]), también conocido como idic(15) o tetrasomía del 15 (OMIM 608636), es una entidad clínica claramente definida que presenta hipotonía central, retardo del desarrollo, compromiso intelectual, epilepsia, comportamiento autista y algunas anomalías menores en su fenotipo, su incidencia estimada es de 1 caso por cada 30.000 nacimientos, afecta por igual ambos sexos1. Desde el punto de vista citogenético se definió como una duplicación invertida de la zona comprendida entre la región terminal del brazo corto hasta la región 12 a 13 del brazo largo del cromosoma 15 (invdup[15][pter→q12-13::q12- 13→pter]), incluyendo la región q11 (q11→q13) donde se encuentran la regiones críticas de los síndromes de Prader Willi (PWS) y Angelman (AS). En la mayoría de los casos estudiados hasta la fecha, el cromosoma extra presente es de origen materno, relacionándose a su vez con anomalías fenotípicas y con edad materna avanzada2–4.
La región cromosómica 15q11q13, conocida por inestabilidad, secundaria a la presencia de elementos repetitivos de ADN, es altamente susceptible a la formación de rearreglos clínicamente relevantes, como la formación de marcadores supernumerarios formados por la duplicación invertida del cromosoma 15, la cual puede resultar en tetrasomía del brazo corto del cromosoma 15 (15p) o tetrasomía parcial del brazo largo del cromosoma 15 (15q) 5. La variabilidad fenotípica de estos pacientes suele estar dada por la región específica involucrada en cada rearreglo cromosómico. Reportamos un caso del síndrome de tetrasomía del 15 con múltiples anomalías físicas y epilepsia, que involucra a 15q11.2 como punto de corte.
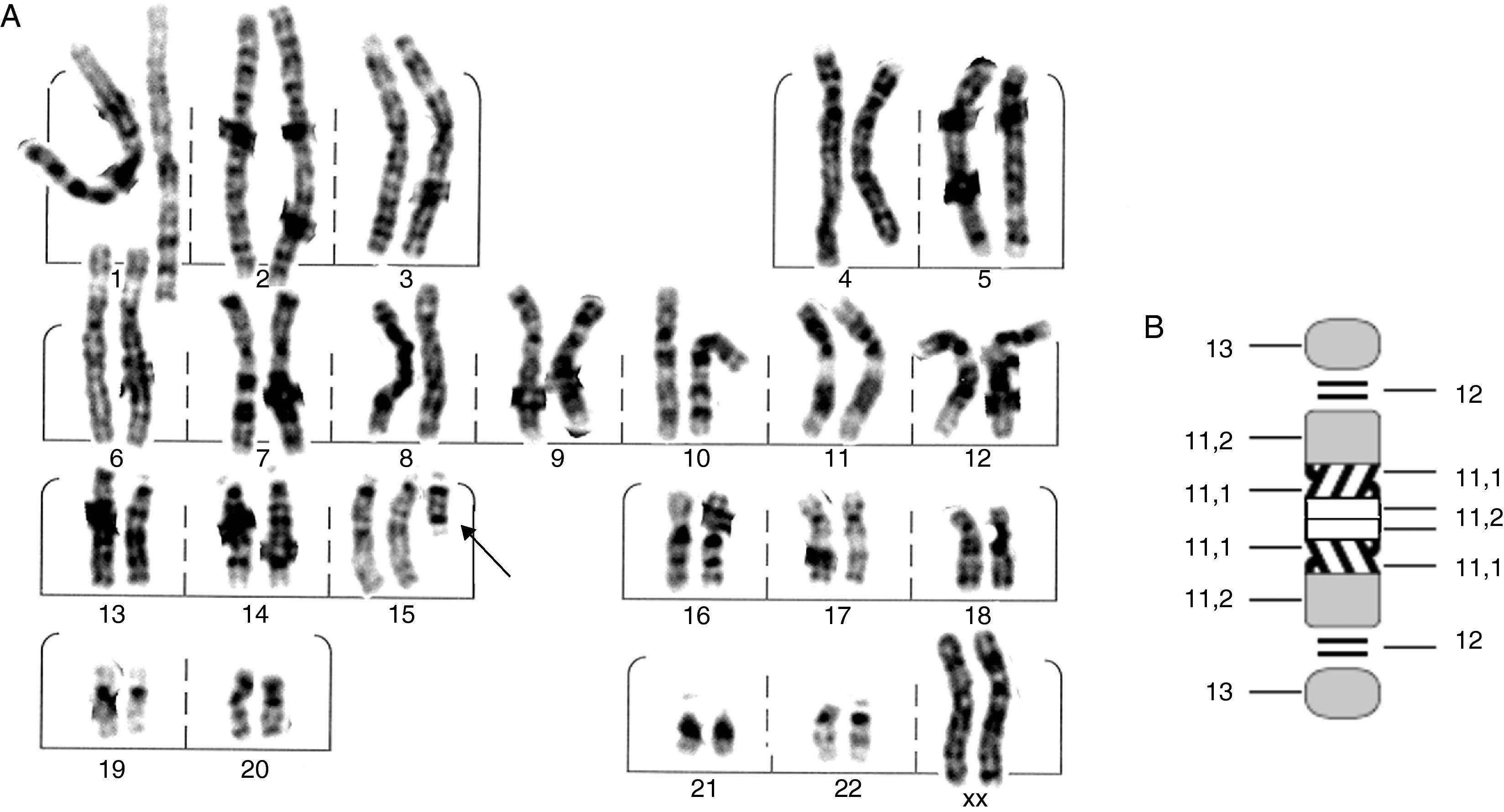
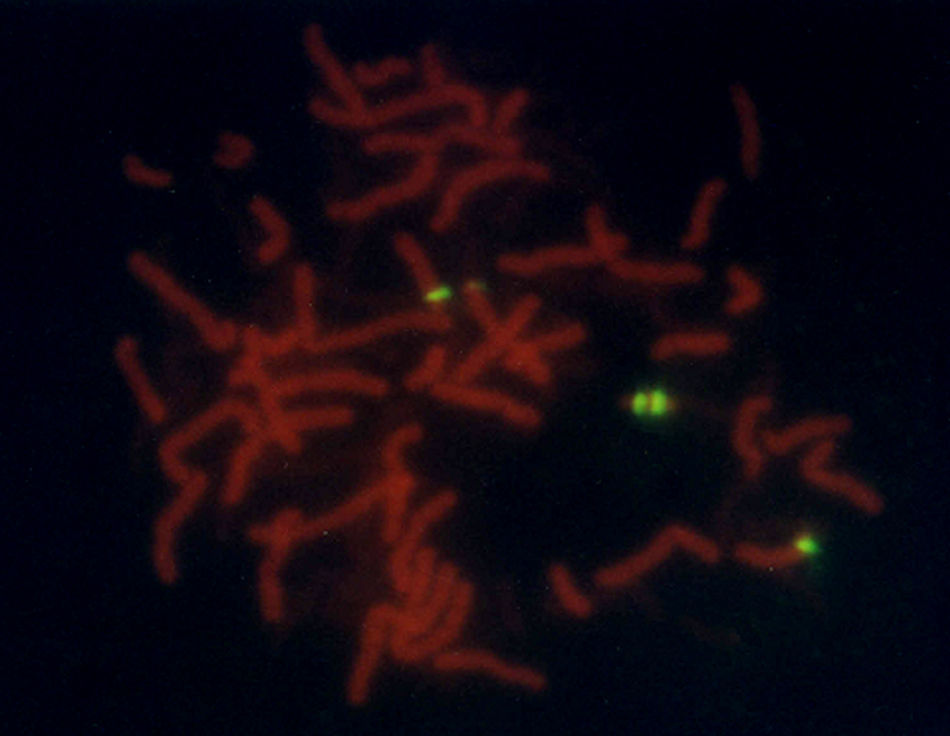
Se trata de una paciente femenina remitida a la consulta de genética con diagnóstico de síndrome convulsivo en estudio, acompañado de anomalías menores. Al examen físico presentó facies ovaladas, nariz voluminosa, fisuras palpebrales ligeramente oblicuas, pabellones auriculares alados, hipoplasia mediofacial moderada, micrognatia leve, extremidades alargadas, tendencia al genu-valgo y pie equino-varo. Se realizaron pruebas bioquímicas, encontrándose aminoácidos normales; en el cariotipo se determinó la presencia de un minicromosoma que pudiera corresponder a parte de un cromosoma 14 o 15; por los hallazgos fenotípicos se reportó como una trisomía parcial del cromosoma 15 (15pter→15q1:) (fig. 1). Para confirmar este hallazgo, se realizó FISH con sonda centromérica del cromosoma 15; tanto en núcleos como en cromosomas 15, se observaron cuatro puntos fluorescentes, uno en cada cromosoma 15 normal y dos de ellos sobre el minicromosoma, lo que indica la presencia de 2 centrómeros de cromosoma 15, (fig. 2) interpretándose como una tetrasomía parcial de 15q, resultado de una duplicación invertida del segmento 15pter → 15q11.2: 47,XX,+der(15)(pter→q1:).ish invdup(15)(pterq11.2)(D15Z1++) o 47,XX,+der(15)(pter→q1:).ish invdup(15)(pter→q11.2::q11.2→pter)(D15Z1++).
; B. Ideograma del cromosoma adicional, especificando puntos de corte.")

En general, los pacientes con tetrasomia del 15 presentan retardo del desarrollo con sedestación entre los 10 a 20 meses de edad y deambulación entre los 2 y 3 años, al igual que déficit cognitivo en las áreas del lenguaje expresivo (lenguaje ecolálico) y comprensión. Su comportamiento a menudo es descrito como autista-like o autista. La epilepsia es de características variadas, sin predominancia de un tipo específico, a excepción del síndrome de Lennox-Gastaut en cuatro pacientes descritos por Battaglia en 1997, quienes presentaban convulsiones tónicas/atónicas, tónico-clónicas y ausencias; otros tipos de crisis descritas son las complejas parciales, mioclónicas generalizadas de inicio en la adultez, tónico-clónicas generalizadas y epilepsia benigna con descargas centro-temporales, con un rango de presentación que varía entre los 6 meses hasta los 9 años. Se ha sugerido que el grado de compromiso mental y retardo psicomotor pueden estar correlacionados con la severidad de las convulsiones y su intratabilidad, sin embargo, poco estudios han contribuido a dilucidar este aspecto2. En general, las facies de los pacientes son normales; pueden encontrarse hallazgos inespecíficos como fisuras inclinadas hacia abajo, pliegue epicántico, ojos profundos, pabellones de baja implantación y/o rotados posteriormente, paladar alto, nariz ancha, narinas antevertidas, clinodactilia del quinto dedo en manos, sindactilia parcial del segundo y tercer dedo en pies3. Nuestro paciente no presentó ninguna de las alteraciones neurológicas descritas a excepción de convulsiones y anomalías físicas menores, junto con otras más importantes como pie equino-varo.
El cromosoma 15, más específicamente el locus 15q11-q13, es un clúster de genes de impronta génica esencial para el neurodesarrollo normal en los mamíferos. Como se mencionó antes, este locus presenta gran inestabilidad, específicamente a nivel de cinco puntos comunes de ruptura (De BP1 aBP5,) que pueden ocasionar rearreglos genómicos incluyendo deleciones y duplicaciones. Uno de los mecanismos sugeridos para la formación de la invdup(15) involucra la recombinación tipo-U entre cromosomas homólogos, seguida de no disyunción e inactivación de uno de los centrómeros. La forma más frecuente de idic(15) se caracteriza por un evento de recombinación asimétrica entre BP4 y BP5; esto conlleva a tetrasomía del intervalo entre el centrómero y BP4 y trisomía del intervalo entre BP4 y BP53. Las duplicaciones maternas que ocurren como duplicaciones intersticiales y los cromosomas isodicéntricos supernumerarios llevan a desórdenes variables del neurodesarrollo con características autísticas en algunos casos; esta duplicación es la principal causa citogenética de autismo, produciendo el 1-3% de los casos6. De igual manera se sabe qué factores ambientales y genéticos adicionales influencian la variabilidad final del cuadro clínico al igual que en la mayoría de los síndromes comportamentales, que se han visto relacionados con un diverso grupo de enfermedades infecciosas como la embriopatía por rubeola y encefalitis7. Cabe recordar que la segunda causa de tetrasomía del cromosoma 15 es la triplicación intracromosómica; se sugiere que este tipo de anomalía surge secundaria a un proceso de dos pasos durante la meiosis, inicialmente ocurre intercambio tipo-U (contrario al intercambio normal, tipo-X), resultando en una duplicación invertida dicéntrica, posteriormente se recombina con un cromosoma 15 normal y forma la triplicación, o también se puede formar como resultado de un intercambio tipo-U entre tres cromátides en un solo paso8,9.
El hecho que la gran mayoría de duplicaciones del cromosoma 15 (dup[15]) y de SMC(15) que contienen la región critica de PWS/AS sean de origen materno implican que la duplicación de origen paterno o es poco frecuente y letal, por lo que raramente sobrevive el embarazo a término o no tiene efecto fenotípico y pasa desapercibida. Sin embargo, no se puede descartar que la aparente ausencia del fenotipo es secundario a un efecto de posición que consistiría en que el gen o genes transcripcionalmente activos proximales al punto de rotura en 15q, se inactiven10,11.
Gracias a las técnicas actuales de citogenética básica y molecular, se ha podido establecer la correlación entre el tamaño del fragmento involucrado en la duplicación y el fenotipo. Individuos con una región de duplicación pequeña y muy cercana al centrómero son completamente sanos, mientras que duplicaciones que comprometen regiones más grandes con puntos de rotura en q12 y q13 ocasionan retardo mental, autismo o convulsiones1. En este caso en particular, la región comprometida 15q11.2, incluye genes de importancia potencial en la epilepsia como son los genes de las subunidades del receptor gamma-aminobutírico (GABA), conocido como el principal neutrotransmisor inhibitorio en el cerebro de los mamíferos, pero a su vez, también se ha demostrado una propiedad excitatoria a nivel de las neuronas maduras del núcleo supraquiasmático del hipotálamo. Esta actividad dual puede estar regulada por las oscilaciones diurnas de las concentraciones de cloro intracelular. Por lo tanto, la duplicación y expresión excesiva de estos receptores de neutrotransmisores puede contribuir a la presencia de la enfermedad, tal como se ha observado en adultos con epilepsia e inv dup(15) al igual que hiperactividad y agresividad5,11,12.
Puesto que la mayoría de estos pacientes al examen físico no presentan dismorfismo o anomalías menores, el estudio cromosómico puede pasarse por alto y por ende, no se diagnostica la anomalía cromosómica base en estos pacientes. Por tal motivo, es recomendable que a pacientes con retardo del desarrollo, epilepsia (principalmente de difícil manejo) y autismo, que presenten o no anomalías dismórficas al examen, se les realice cariotipo de alta resolución y citogenética molecular para descartar este tipo de alteraciones cromosómicas.
Se reporta este caso con fines académico-pedagógicos dada la baja incidencia de esta patología (1:30.000 nacidos vivos) y la importancia del estudio complementario que fue realizado en esta paciente, lo que finalmente permitió llegar a un diagnóstico definitivo y por ende, ofrecer una correcta y apropiada asesoría genética a los padres.
El presente artículo fue presentado en formato Poster, en el XI Congreso Colombiano de Genética Humana. Medellín. Octubre 2010.

