



Guillain-Barré syndrome (GBS) is a heterogenous condition and may be mimicked by other acute polyneuropathies. Both acute porphyria and systemic lupus erythematosus (SLE) are included in this differential diagnosis. We report an instance of acute polyneuropathy in a pregnant patient with known SLE, whose neurological picture occurred during a flare of SLE but was found to be due to a previously undiagnosed hereditary coproporphyria (HCP).
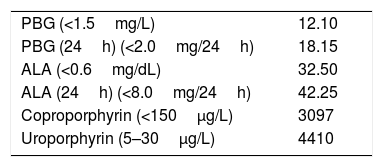
We present the case of a 25-year-old Asian female with SLE with severe multisystemic involvement, diagnosed during a life-threatening flare in her first pregnancy. There was no other relevant personal or family history. Medication included prednisolone 5mg, azathioprine 100mg, and hydroxychloroquine 400mg. She was admitted for abdominal pain, vomiting, and constipation, followed by dark urine; she also described other self-limiting abdominal pain episodes over the previous 2 years. On examination, persistent tachycardia was noted. Laboratory studies were significant for hypocomplementemia, elevated erythrocyte sedimentation rate and elevated β-Human chorionic gonadotropin compatible with a 1–2-week pregnancy. There was no laboratory evidence of hematuria. Abdominal and obstetric ultrasound, and abdominal MRI were unremarkable. A provisional diagnosis of SLE-associated autonomic neuropathy was made and she was treated with IV methylprednisolone pulse plus an increase in prednisolone to 1mg/kg/day. A rapidly progressive quadriparesis developed the first week after admission. On day 7 the patient was unable to independently walk, had a generalized areflexia, and patchy hypoesthesia. Electrodiagnostic studies showed increased F wave latency and absent H reflexes, suggesting GBS. CSF studies were normal. Despite treatment with intravenous immunoglobulin, followed by plasma exchange, her neurological condition continued to deteriorate, with facial diplegia, worsening of quadriparesis (grade 2 MRC in the proximal segments and grade 0 distally), marked hypopalesthesia and proprioceptive errors up to the knees and elbows, and periods of severe dysautonomia. Repeat nerve conduction studies showed an acute sensory-motor axonal neuropathy (AMSAN). Meanwhile, the patient developed thrombocytopenia, prolonged clotting times, hypoproliferative anemia and elevated hepatic transaminases, and a provisional diagnosis of SLE flare with SLE-related acute polyneuropathy was made. The patient agreed to an interruption of the pregnancy, followed by treatment with cyclophosphamide. Systemic manifestations resolved, without any improvement of neurological symptoms. Additional investigations for GBS mimics were ordered, and urinary porphobilinogen and porphyrins were markedly elevated (Table 1). After withdrawal of potentially porphyrinogenic drugs and administration of IV hemin, the patient showed dramatic improvement, with resolution of sensory manifestations and plateauing of muscle strength. She was discharged to a rehabilitation center. At 20 months after onset, full recovery had occurred except for bilateral grade 4 MRCankle dorsiflexion. Genetic testing revealed a previously unreported CPOX gene variant (NM_000097.5:c.245T>C [p.Leu82Pro]).
The present case illustrates a challenging differential diagnosis of acute polyneuropathy. Our patient had both an established diagnosis of SLE and laboratory evidence of increased disease activity. Still, she defied Occam's razor and final diagnosis was of a second chronic disease, hereditary coproporphyria (HCP), as the cause of the neurological symptoms.
Clinical reasoning favors acute porphyric neuropathy against SLE-associated neuropathy: there had been self-limiting abdominal pain episodes accompanied by dark urine, abdominal pain and dysautonomia occurred before the acute neuropathy, and response to immunosuppression was absent while there was a response to hemin treatment and withdrawal of porphyrinogenic drugs. The lack of cutaneous manifestations is not unexpected, since they are far less frequent than neurovisceral manifestations in patients with HCP, occurring only in 5–30% of patients.1 The main limitations of the present report include limited biochemical testing (unavailable fecal and plasma porphyrin testing; urinary porphyrins and PBG unadjusted for creatinine) and incomplete genetic testing, namely of unaffected family members. However, clinical presentation was consistent with neurovisceral porphyria, no alternative causes were present for the expressive elevation of PBG and porphyrins, and a CPOX variant was found.
To the best of our knowledge, two other cases of SLE and hereditary coproporphyria have been reported.2,3 This association is most likely fortuitous, considering the low number of reports and weak evidence for shared pathological mechanisms. On the other hand, both entities present mainly in women of childbearing age, and can be precipitated by external factors. In the present case, pregnancy was likely the common factor precipitating flares of both SLE and HCP, although it is an infrequent precipitant for the latter.4 An SLE flare could precipitate a porphyric attack through caloric deprivation, but the presenting abdominal symptoms and dysautonomia were likely porphyric. In addition, drugs used to treat SLE could have aggravated the porphyria, since methylprednisolone, cyclophosphamide, and hydroxychloroquine are all possibly porphyrinogenic.5
Though acute porphyrias are rare disorders, one study found a surprisingly high prevalence of 11% previously undiagnosed cases in patients with acute polyneuropathy or encephalopathy in combination with abdominal pain or dysautonomic features.6
Our case highlights the necessity of including porphyria in the differential diagnosis of an acute polyneuropathy, even when alternative causes are present. The availability of specific therapy and preventive measures has important clinical implications, not only for the patient but also to family members, since without prompt treatment recovery is unpredictable and often incomplete.
Conflict of interestThe authors declare no conflicts of interest and no disclosures relevant to the manuscript. Informed consent was obtained from the patient for publication of this case report.
The authors wish to thank Dr. Estela Nogueira, from the Nephrology Department, and Dr. Catarina Resende, from the Rheumatology Department, for their expertise and assistance in the present case.
