



La angiopatía amiloide cerebral (AAC) se caracteriza por el depósito de beta-amiloide en la media y adventicia de los vasos corticales y leptomeníngeos1. Favorece una disrupción en su arquitectura ocasionando una rotura vascular2, siendo la hemorragia parenquimatosa lobar (única o múltiple) su presentación más frecuente; también como focalidad neurológica transitoria3 y deterioro cognitivo. La presencia de múltiples microsangrados córtico-subcorticales en imagen RMN eco gradiente o de susceptibilidad magnética (SWI) es un signo radiológico sugestivo de vasculopatía amiloide. Las imágenes por tomografía por emisión de protones (PET) con ligandos de amiloide pueden ser útiles en la detección de la AAC, sin discriminar el depósito vascular (propio de la AAC) del parenquimatoso (característico de la enfermedad de Alzheimer [EA])4.
El riesgo de AAC se incrementa con la edad, detectándose hasta en el 38% de los pacientes entre 80-89 años y hasta en el 42% de los pacientes por encima de 90 años5. Su existencia es rara en sujetos menores de 50 años.
La AAC se puede establecer en ciertas localizaciones de forma preferente, como la región parieto-temporal, siendo la arteria cerebral media la más implicada. Sin embargo, la manifestación clínica unilateral es excepcional y se desconoce su justificación5,6. Una hipótesis es que corresponda a un estado evolutivo de la AAC, otra es que responda a factores precipitantes que favorezcan el depósito de amiloide en un patrón lateralizado.
Aunque la «siembra experimental» de beta-amiloide es un fenómeno bien conocido7,8, la transmisión amiloidea en humanos no ha sido reportada hasta muy recientemente. Se postula un mecanismo prion-like similar a la enfermedad de Creutzfeldt-Jakob iatrogénica8–10.
Describimos el caso de un paciente con antecedente de neurocirugía en la juventud, que presenta hematomas lobares de repetición sugestivos de angiopatía amiloide.
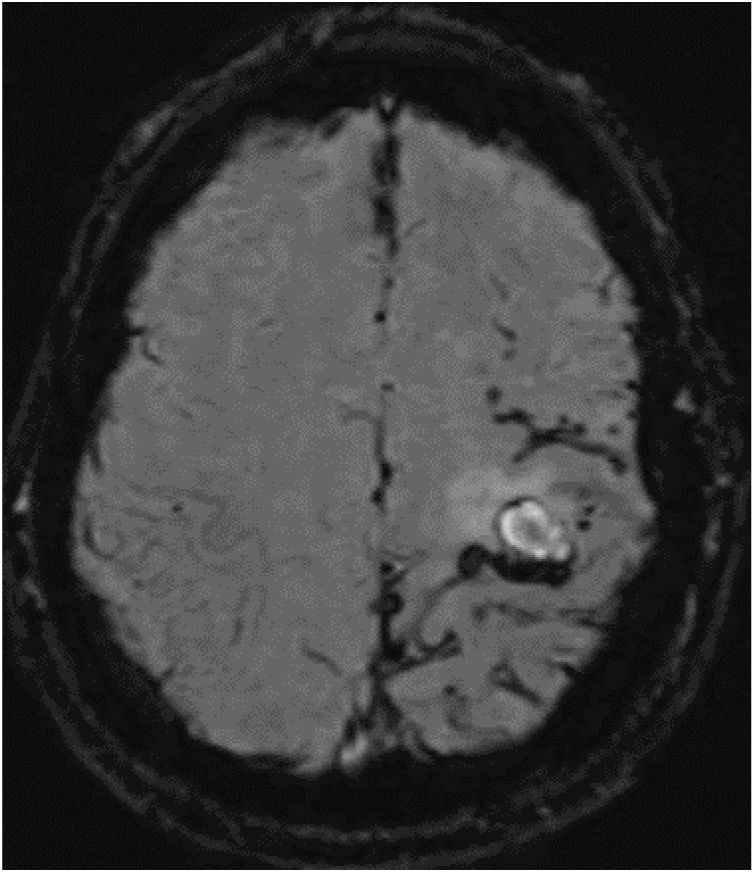
Varón de 52 años, caucásico, sin antecedentes familiares de enfermedades neurodegenerativas. Hábito tabáquico y síndrome de apnea del sueño. Intervenido a los 18 años de un granuloma eosinófilo en región parietal izquierda, sin radioterapia posterior. Fue estudiado a los 44 años por quejas cognitivas, con estudio neuropsicológico, sin alteraciones. La tomografía computarizada por emisión de fotón único (SPECT) describió una hipoperfusión cortical en temporal posterior izquierdo y contralateral. A los 50 años consulta por episodios autolimitados de disestesia en brazo y hemifacies derecha. Ante la sospecha de crisis sintomáticas remotas, se inicia tratamiento con acetato de eslicarbacepina. La RMN cerebral de control evidencia cambios isquémicos crónicos de predominio izquierdo; sin alteraciones en la circulación mediante ecografía Doppler. Es valorado a los 52 años por presentar debilidad brusca en su mano derecha. La TC cerebral muestra un pequeño hematoma subagudo parenquimatoso en la convexidad frontal izquierda. En la secuencia SWI de la RMN cerebral (fig. 1) se aprecian numerosos focos de sangrado en surcos de la convexidad frontoparietal izquierda en relación con siderosis leptomeníngea.

RMN cerebral y SWI. Se aprecian numerosos focos puntiformes de sangrado en lóbulo parietal, en menor cantidad frontal posterior y alguno aislado en lóbulo occipital, todos en el lado izquierdo, así como fino ribete de hiposeñal en espacio subaracnoideo a nivel de cisura de Rolando en relación con siderosis leptomeníngea.
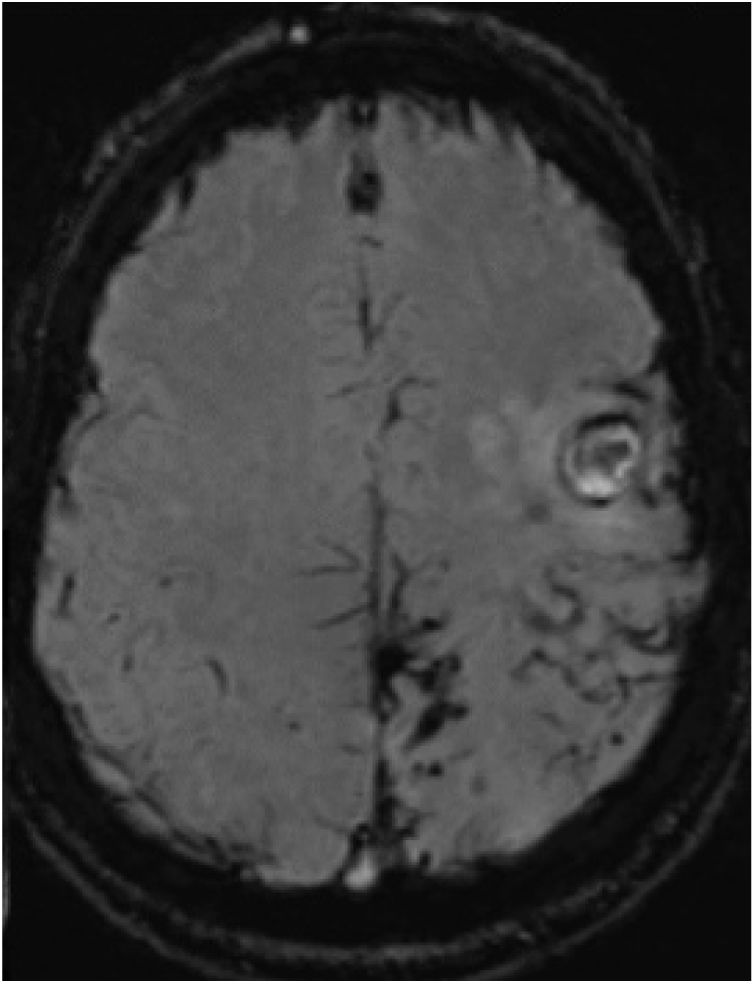
Tres meses después, presentó un empeoramiento brusco de su secuela motora, apareciendo en la TC cerebral 2 focos hemorrágicos corticales frontales izquierdos agudos. En la consiguiente RMN con secuencias SWI (fig. 2), persisten depósitos de hemosiderina en lóbulos parietal, frontal posterior y occipital izquierdos, similares a la RMN previa, sin visualizarse ninguno en hemisferio derecho. No había realce significativo tras la administración de gadolinio. Las secuencias vasculares no aportaron datos significativos. El estudio de coagulabilidad y de autoinmunidad fue normal.

Se realizó PET amiloide con 18F-flutemetamol, siendo el análisis visual positivo de forma bilateral y difusa en todo el córtex. No se analizaron biomarcadores en LCR ni genotipado de la ApoE.
Mediante exoma clínico se estudiaron 6.102 genes. Sin presentar mutaciones en los asociados con la AAC hereditaria como APP, PSEN1, PSEN2, ITM2B (membrana integral proteína 2A o BRI2) y cistatina 3 (CST3).
En el estudio neuropsicológico posterior no se ha detectado deterioro cognitivo. Presenta dificultades moderadas en dominios visoespaciales que se consideran secundarios a la lesión estratégica parietal.
Nuestro paciente cumpliría los criterios de Boston modificado para la probable AAC4, con la excepción del inicio previo a los 55 años. La imagen de siderosis podría justificarse por el propio procedimiento quirúrgico, pero no explicaría los hallazgos en la PET amiloide ni los hematomas lobares de repetición.
En individuos jóvenes (30-57 años) con inicio precoz de AAC, se ha descrito un antecedente neuroquirúrgico cerebral (con o sin injerto de duramadre), espinal u otro proceso invasivo (como embolización de carótida externa con extractos de duramadre) precedido en al menos 3 décadas11–14. Los casos descritos presentan clínica e imagen radiológica característica de ACC, con afectación predominante del hemisferio ipsilateral a la cirugía y depósito amiloide generalizado.
Si bien la prevalencia es desconocida, nuestro caso apoya la posible transmisión amiloidea a través de procedimiento neuroquirúrgico9,15.
El consentimiento fue obtenido verbalmente a través de contacto telefónico dado la restricción de visitas presenciales por la pandemia.
FinanciaciónLa presente investigación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.

