



The DYNC1H1 gene, located on chromosome 14q32,1 encodes the cytoplasmic dynein heavy chain, part of a motor complex with multiple subunits that plays an essential role in retrograde axonal transport and other intracellular functions.2
DYNC1H1 mutations are associated with a broad spectrum of diseases, including intellectual disability, epileptic encephalopathy, malformations of cortical development, Charcot-Marie-Tooth disease, hereditary spastic paraplegia, microcephaly, and spinal muscular atrophy with lower limb involvement.1–5
We present the case of an 8-year-old boy with a de novo missense mutation of DYNC1H1, who was clinically diagnosed with spinal muscular atrophy (predominantly affecting the lower limbs) and predominantly inattentive attention-deficit/hyperactivity disorder.
The patient was the son of young, healthy, non-consanguineous parents. The reason for consultation was inattention and excess motor activity, mainly affecting academic performance and peer relationships. Pregnancy and delivery were uneventful. He had personal history of generalised hypotonia; psychomotor developmental delay, with free ambulation at 30 months and good language development; suspected ataxia since 36 months of age; coeliac disease; and eosinophilic oesophagitis. Regarding family history, his maternal grandmother presented amyotrophic lateral sclerosis. Physical examination detected no pigmentation alterations or dysmorphic features; he presented generalised hypotonia with wide-based gait and predominantly lower-limb weakness. Stretch reflexes were hypoactive and plantar reflex was flexor. The patient did not present dysmetria.
When the patient was 3 years old, he underwent a genetic study with a targeted panel for ataxias, as well as an electromyography and electroneurography study; all results were normal. A brain and spinal MRI study revealed reduced thickness of the right parasagittal anterior cranial fossa with adaptation of the right inferior frontal and olfactory gyri and the right uncinate fasciculus.
Due to the patient’s academic difficulties, at the age of 6 years he underwent a neuropsychological evaluation, which revealed a total IQ of 85 in the Wechsler Intelligence Scale for Children-V, scoring low (5 points) for the digit span; in continued execution tasks, he presented high scores for errors of omission and variable attention (higher than percentiles 95-99). His score for manual dexterity in the Movement Assessment Battery for Children-2 was in the first percentile. Clinical records completed by the patient’s parents and teachers were consistent with the neuropsychological evaluation results, indicating pronounced attentional difficulties.
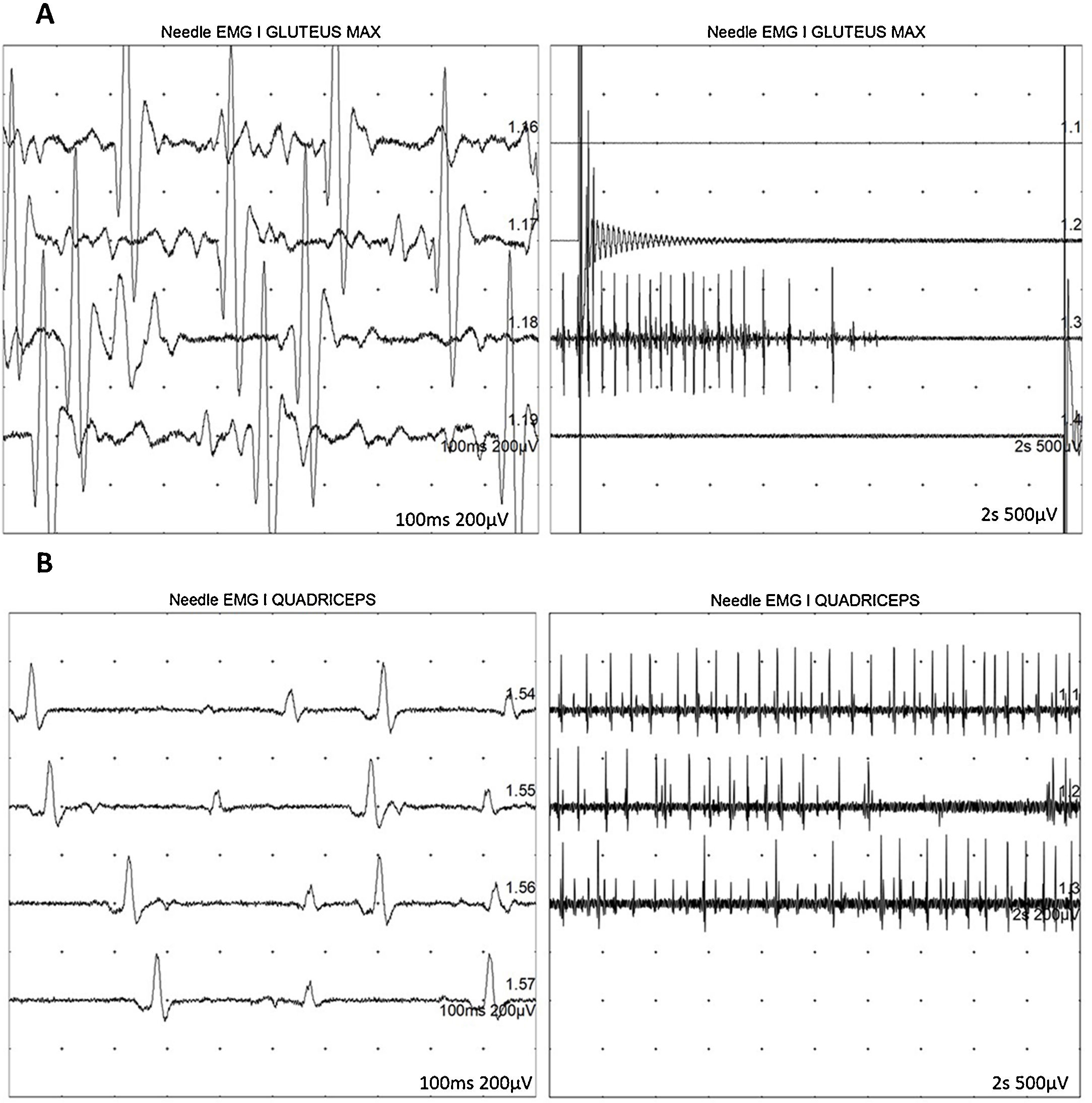
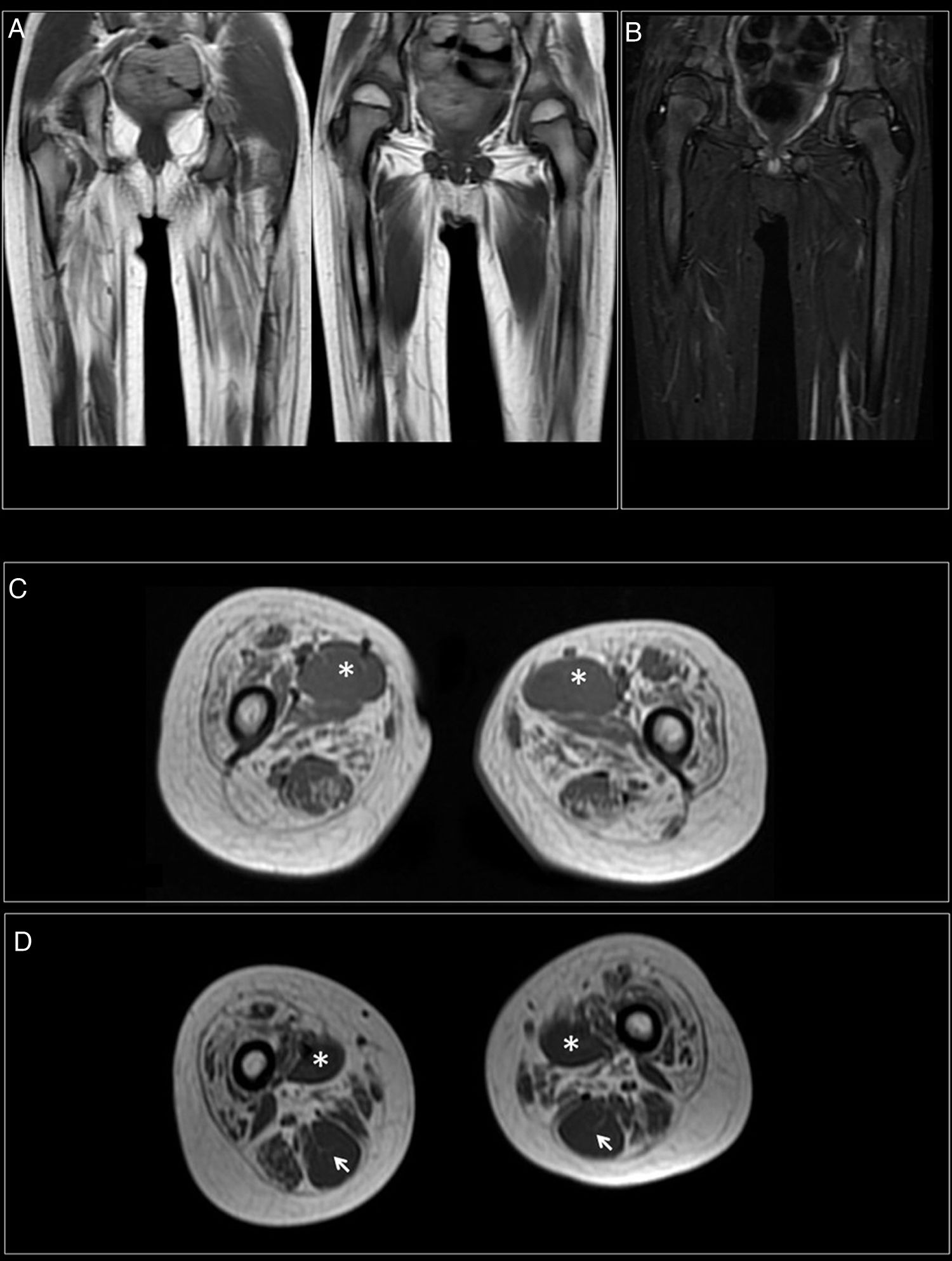
At 7-and-a-half years old, when he consulted at our department, the genetic study was expanded with trio whole exome sequencing, which revealed a DYNC1H1 mutation (c.751C>T; p.Arg251Cys); we repeated the neurophysiological study (Fig. 1), detecting severe, stable, chronic, axonal neurogenic changes in proximal regions of the lower limbs (gluteal/crural muscles). We also performed an MRI study of the lower limbs (Fig. 2), which revealed complete fatty replacement of the proximal muscles in both legs; symmetrical bilateral involvement was observed on coronal T1-weighted sequences, whereas T2-weighted sequences showed no signs of acute denervation.
 and quadriceps muscles (B) show increased motor unit potential duration and decreased recruitment, indicating loss of motor units.")
Neurophysiological study showing severe, chronic, axonal neurogenic changes in proximal regions of the lower limbs. Electromyography studies of the left gluteus maximus (A) and quadriceps muscles (B) show increased motor unit potential duration and decreased recruitment, indicating loss of motor units.
 Coronal T1-weighted sequence showing fatty replacement of the proximal muscles of both lower limbs; involvement is symmetrical and bilateral. (B) T2-weighted sequences showed no signs of acute denervation. (C) Symmetrical fatty replacement, with particular involvement of the anterior (quadriceps) and medial (adductor magnus) compartments. The adductor longus is preserved (asterisk). (D) Partial fatty replacement of the ischiotibial muscles in the posterior compartment, with the exception of the semitendinosus (arrow).")
Muscle MRI. (A) Coronal T1-weighted sequence showing fatty replacement of the proximal muscles of both lower limbs; involvement is symmetrical and bilateral. (B) T2-weighted sequences showed no signs of acute denervation. (C) Symmetrical fatty replacement, with particular involvement of the anterior (quadriceps) and medial (adductor magnus) compartments. The adductor longus is preserved (asterisk). (D) Partial fatty replacement of the ischiotibial muscles in the posterior compartment, with the exception of the semitendinosus (arrow).
Our patient’s clinical phenotype is similar to those previously described in the literature.6 Spinal muscular atrophy is a group of hereditary diseases that cause progressive muscle degeneration and weakness secondary to the loss of spinal or bulbar motor neurons. Most cases are associated with mutations in the SMN1 gene.7 In recent reports, cases have been associated with such other genes as UBA1, DYNC1H1, and BICD2.4,8,9 With regard to the DYNC1H1 gene, an article from 2018 by Chan et al.6 reported 4 unrelated patients with the missense mutation c.751C>T in heterozygosis, with a phenotype coinciding with that observed in our patient: predominantly lower-limb muscle weakness, learning difficulties in 2 cases and intellectual disability in the other 2, mild brain MRI alterations, and evidence of spinal muscular atrophy in lower-limb MRI studies. The authors also note that muscle MRI (a non-invasive test) is more specific than muscle biopsy in the diagnosis of this disease.6 In 2012, Tsurusaki et al.4 described a different DYNC1H1 mutation (c.917A>G) in 2 patients with spinal muscular atrophy. Cognitive problems have also been reported; we recommend the article by Fiorillo et al.,10 who describe attentional problems similar to those of our patient. Initially, mutations in the first domains of this gene were associated with lower motor neuron involvement, whereas mutations at the N-terminal end were associated with cognitive problems or malformations of cortical development4,11,12; however, more recent studies and detailed analysis of international data have raised doubts about this genotype-phenotype correlation.13 The study by Chan et al.6 demonstrates how an identical mutation in the tail domain of DYNC1H1 can indistinctly be associated with intellectual disability or learning difficulties. Similarly, mutations in the tail or motor domains have been associated with malformations of cortical development. Thus, phenotypic variability may be influenced by the domain affected, but conditioned by other genetic or even environmental factors.
This case underscores the importance of whole-genome sequencing techniques, which offer greater diagnostic yield than panels or arrays and are recommended as the first-line study in current guidelines; trio sequencing (proband plus parents) further increases sensitivity.14 We would stress that the combination of both techniques may constitute a potential tool for individuals with undiagnosed neurodevelopmental disorders,15 as well as the importance of properly interpreting genetic study results. The diagnostic yield and value of these studies are known to increase when they are interpreted in the hospital context by professionals with access to the patient and to the results of complementary studies.16
FundingNone.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Fernández Perrone AL, Moreno Fernández P, Álvarez S, Fernández-Jaén A. Mutación de novo en DYNC1H1, atrofia muscular espinal y problemas atencionales. Neurología. 2022;37:406–409.
