



El gen DYNC1H1 está localizado en el locus 14q321. La cadena pesada de dineína citoplasmática es parte de un complejo motor, con múltiples subunidades, esencial en el transporte axonal retrógrado y otras funciones intracelulares2.
Las mutaciones del DYNC1H1 se asocian a un amplio espectro de patologías: discapacidad intelectual, encefalopatía epiléptica, malformaciones corticales, Charcot-Marie-Tooth, paraplejía espástica hereditaria, microcefalia y atrofia muscular espinal con afectación de extremidades inferiores1–5.
Presentamos el caso de un varón de 8 años en el que se identifica una variante missense, de novo, en el gen DYNC1H1, con diagnóstico clínico de atrofia muscular espinal (predominante en extremidades inferiores) y trastorno por déficit de atención/hiperactividad- presentación predominantemente inatenta.
El caso es hijo de padres sanos, jóvenes, no consanguíneos. El motivo de consulta es inatención y un exceso de actividad motora que afecta principalmente el entorno académico y las relaciones con el grupo de pares. Producto de un embarazo y parto sin datos relevantes. Como antecedentes personales destaca hipotonía generalizada, desarrollo psicomotor lento, con marcha liberada a los 30 meses y buen desarrollo del lenguaje; sospecha de ataxia desde los 36 meses; celiaquía y esofagitis eosinofílica. De los antecedentes familiares cabe mencionar la abuela materna con esclerosis lateral amiotrófica. En la exploración física no se observan alteraciones de la pigmentación ni rasgos dismórficos; destaca hipotonía generalizada con marcha con aumento de base de sustentación y debilidad de predominio en extremidades inferiores. Los reflejos miotáticos son hipoactivos con reflejo plantar flexor presente. No dismetría.
A la edad de 3 años se realizó estudio genético con panel dirigido a ataxias, electromiograma y electroneurograma, todos con resultados normales. En la resonancia magnética cerebral y medular se identifica mayor excavación ósea de la fosa craneal anterior parasagital derecha con adaptación de los giros frontales inferiores y olfatorios derechos, así como del fascículo uncinado derecho.
Dadas las dificultades escolares, a la edad de 6 años se realiza evaluación neuropsicológica que revela un CIT de 85 en el test WISC-V, destacando a la baja, la puntuación 5 en dígitos; en tareas de ejecución continuada se registran puntuaciones en errores por omisión y variabilidad atencional elevadas (>pc95-99), y en la batería de evaluación del movimiento para niños (MABC2), destreza en pc 1. Los registros clínicos cumplimentados por padres y profesores, coherentes con la evaluación señalada, son acordes a la presencia de marcados problemas atencionales.
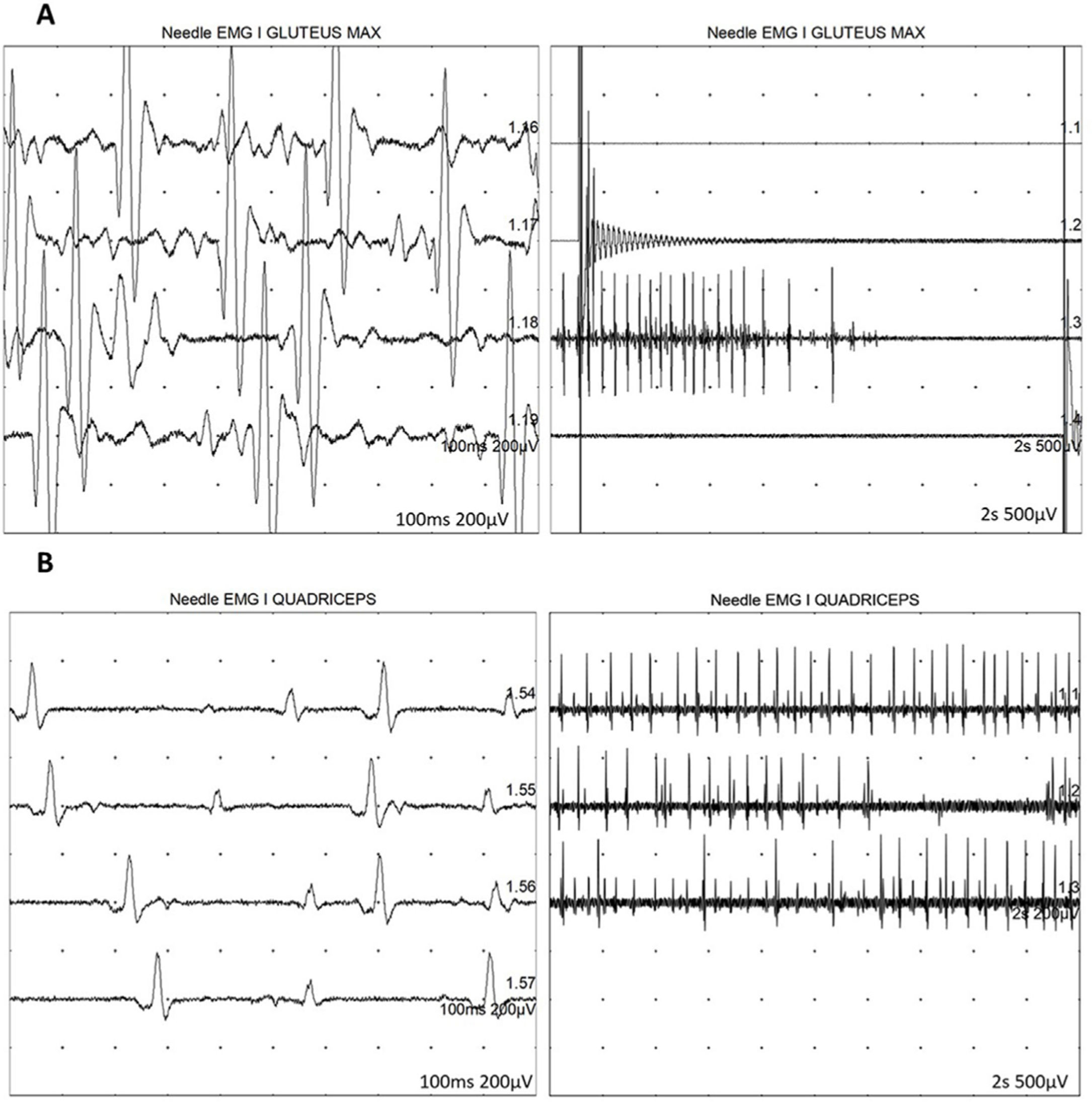
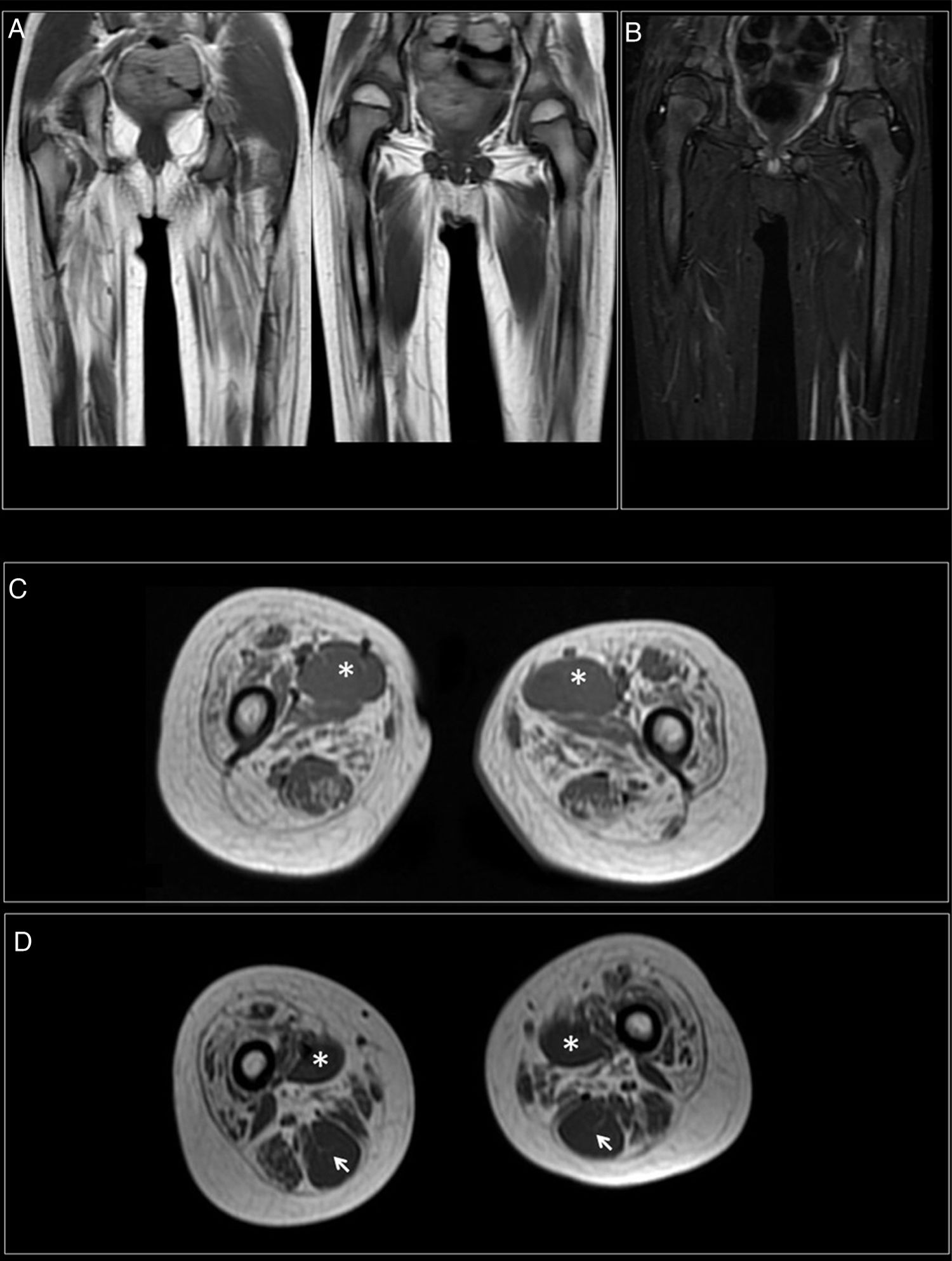
A los 7 años y medio, edad a la que consulta en nuestro servicio, se amplía la evaluación genética con secuenciación exómica masiva en trío que revela mutación en el gen DYNC1H1 (c.751C>T; p.Arg251Cys) y se repite estudio neurofisiológico que describe la presencia de cambios neurógenos crónicos, de carácter axonal, estables, en territorios proximales (musculatura glútea/crural) de extremidades inferiores, grado severo (fig. 1). Además, se realiza resonancia muscular de extremidades inferiores, donde se observa reemplazo graso completo de la musculatura proximal de ambas extremidades inferiores, con afectación simétrica y bilateral en las secuencias potenciadas en T1 en plano coronal, mientras que no se identifican signos de denervación aguda en las secuencias potenciadas en T2 (fig. 2).
 y cuádriceps (B) izquierdos, mostrando una duración aumentada de los potenciales de unidad motora, así como un reclutamiento reducido, expresión de la pérdida de unidades motoras.")
Estudio neurofisiológico con presencia de cambios neurógenos crónicos, de carácter axonal en territorios proximales de extremidades inferiores, grado severo. Se representan EMG de glúteo mayor (A) y cuádriceps (B) izquierdos, mostrando una duración aumentada de los potenciales de unidad motora, así como un reclutamiento reducido, expresión de la pérdida de unidades motoras.
 Reemplazamiento graso completo de la musculatura proximal de ambas extremidades inferiores, con afectación simétrica y bilateral en las secuencias potenciadas en T1 en plano coronal. B) No se identifican signos de denervación aguda en las secuencias potenciadas en T2. C) Reemplazamiento graso simétrico afectando especialmente al compartimento anterior (cuádriceps) y medial (adductor mayor). Preservación del aductor largo (*). D) Reemplazamiento graso parcial de los músculos isquiotibiales del compartimento posterior, a excepción del semitendinoso (flecha).")
RM muscular. A) Reemplazamiento graso completo de la musculatura proximal de ambas extremidades inferiores, con afectación simétrica y bilateral en las secuencias potenciadas en T1 en plano coronal. B) No se identifican signos de denervación aguda en las secuencias potenciadas en T2. C) Reemplazamiento graso simétrico afectando especialmente al compartimento anterior (cuádriceps) y medial (adductor mayor). Preservación del aductor largo (*). D) Reemplazamiento graso parcial de los músculos isquiotibiales del compartimento posterior, a excepción del semitendinoso (flecha).
El fenotipo clínico del caso descrito es similar a los previamente reportados en la literatura6. La atrofia muscular espinal es un grupo de enfermedades musculares hereditarias que causan degeneración y debilidad muscular progresiva secundaria a la pérdida de neuronas motoras espinales o bulbares. La mayoría de los casos se describen asociados a mutaciones en el gen SMN17. Recientemente se han descrito casos asociados a genes como UBA1, DYNC1H1, BICD24,8,9. En relación con el gen comprometido en el caso que presentamos, Chan et al. reportan en 2018 4 pacientes no familiares con mutación en heterocigosis missense en el gen DYNC1H1, c.751C>T, con correlación fenotipo-genotipo que coincide con nuestro paciente: debilidad muscular con predominio en extremidades inferiores, problemas de aprendizaje en 2 de los casos, discapacidad intelectual en los otros 2, anomalías leves en la resonancia cerebral y datos de atrofia muscular espinal en la resonancia muscular de extremidades inferiores; señalan igualmente que la resonancia muscular (prueba no invasiva) es más específica que la biopsia muscular en el diagnóstico de esta patología6. En 2012 Tsurusaki et al. describieron 2 pacientes con clínica de atrofia muscular espinal con una mutación diferente en el gen DYNC1H1 (c.917A>G)4. La presencia de problemas cognitivos asociados también ha sido descrita, aunque destacamos el caso de Fiorillo et al., que describieron la asociación con problemas atencionales como en nuestro caso10. En un primer momento, se asoció la presencia de mutaciones en los primeros dominios de este gen con la afectación de la segunda motoneurona, y la presencia de mutaciones en el extremo N-terminal con problemas cognitivos o malformativo-cerebrales4,11,12; sin embargo, estudios más recientes y el análisis detallado de bases internacionales han desconfigurado esta relación genotipo-fenotipo13. El estudio de Chan et al. demuestra, en el apartado cognitivo, como una idéntica mutación en el dominio de cola del DYNC1H1 puede asociarse a discapacidad intelectual o problemas de aprendizaje indistintamente. En la misma línea, mutaciones en los dominios de cola o motores se han asociado a malformaciones corticales cerebrales. La variabilidad fenotípica, por tanto, puede estar influida por el dominio afectado, pero condicionada por otros factores genéticos o quizás ambientales.
A través del presente caso, subrayamos la importancia de las técnicas de secuenciación genómica masiva; estas pruebas tienen más rentabilidad clínica que los paneles o los estudios por arrays, recomendación de primera línea en las guías actuales, y que utilizando las mencionadas pruebas en trío (probando y sus progenitores) la sensibilidad aumenta14. Es importante tener en cuenta que la combinación de ambas técnicas puede significar una herramienta potencial para individuos con trastornos del neurodesarrollo no diagnosticados15. Cabe recalcar también la importancia de la interpretación correcta de la evaluación genética. Se sabe que la rentabilidad diagnóstica y su utilidad aumenta cuando es interpretada en el contexto hospitalario por personal con acceso al paciente y las evaluaciones complementarias16.
FinanciaciónNinguna.
Conflicto de interesesLos autores no tienen conflictos de intereses en relación con este trabajo.

