




Jansen-de Vries syndrome (JDVS: MIM#617450), also known as “intellectual developmental disorder with gastrointestinal difficulties and high pain threshold (IDDGIP),” is an autosomal dominant disease1,2 described in at least 20 patients1; to our knowledge, the patient presented here is the first case to be diagnosed in Spain. In addition to its extreme rarity, its interest resides in an atypical pattern of obsessive sex drive and the absence of intellectual disability.
The PPM1D gene encodes protein phosphatase Mg2+/Mn2+ dependent 1D, a member of the PP2C family of serine/threonine protein phosphatases. The gene participates in the negative regulation of p53-dependent cellular stress.3 Jansen et al.2 were the first to identify de novo frameshift or truncating mutations in exons 5 and 6 of PPM1D as the cause of a syndrome characterised by a peculiar phenotype, intellectual disability, language and behavioural impairment, gastrointestinal difficulties with cyclic vomiting,2 hypersensitivity to sound, and high pain threshold.2,4
Our patient is an 8-year-old boy who was previously assessed due to mild psychomotor retardation (free ambulation at 2 years) and severe language and communication delay (first words at 4 years).
Harmonious intrauterine growth retardation was observed during pregnancy. The child was born by vaginal delivery at 39 weeks: Apgar score: 9/10; weight: 2.175kg (first percentile); length: 47cm (third percentile); head circumference: 33cm (fifth percentile). We initially observed hypotonia, feeding difficulties, gastro-oesophageal reflux disease, and constipation alternating with intermittent diarrhoea over the years.
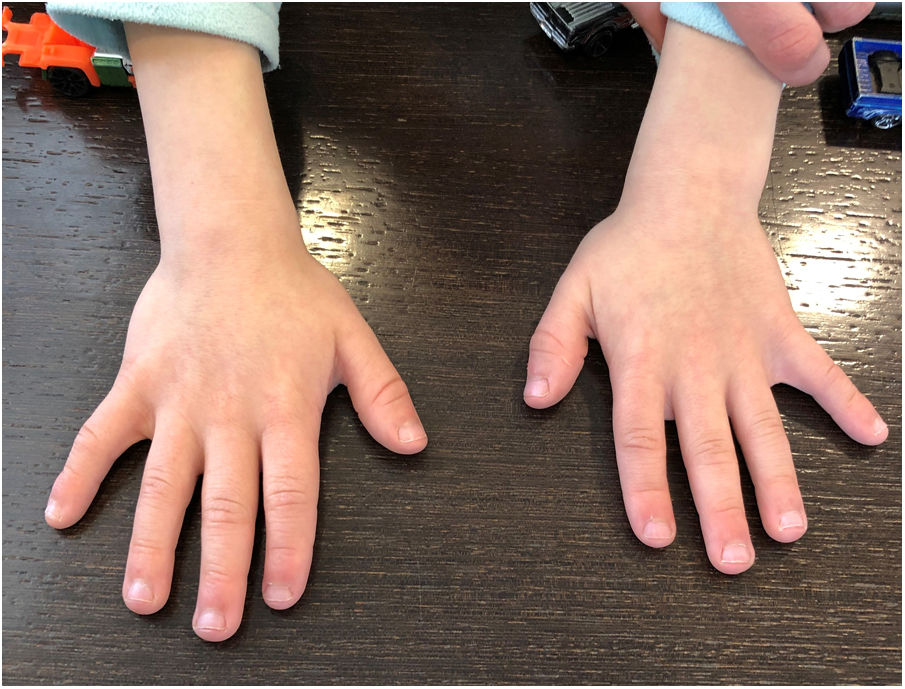
The physical examination performed when the patient was 7 years old revealed a weight of 20.4kg (13th percentile), height of 112cm (second percentile), and head circumference of 48.5cm (third percentile). We also observed subtle dysmorphic features (Fig. 1). The patient also presented lumbar hyperlordosis, small hands with brachydactyly (especially in the distal phalanges) and clinodactyly (Fig. 2), and small feet, with bilateral nail dysplasia. Neurological examination showed no focal signs.
; mild hypotelorism; bilateral epicanthus, predominantly on the left; bulbous nose with small nares; smooth philtrum; thin upper lip and thick lower lip; and large mouth.")
Phenotype of the patient at the age of 7. Wide forehead; low-set, wide ears with backward rotation (the right ear can be seen); mild hypotelorism; bilateral epicanthus, predominantly on the left; bulbous nose with small nares; smooth philtrum; thin upper lip and thick lower lip; and large mouth.
 and clinodactyly.")
His family was concerned by his sexual disinhibition, characterised by a strong desire for social relationships (despite certain difficulties adapting to his classmates) and obsessions with erotic content, associated with compulsive masturbation. He presented a high pain threshold, hypersensitivity to sound, and stereotypy of both hands at the level of the face (to release tension). These repetitive behaviours were not incapacitating, and he was able to make eye contact and showed a good level of empathy. In the neuropsychological evaluation, the WISC-V showed a total intelligence quotient of 84, with a score of 74 in working memory, the most severely impaired item. We observed pronounced attention difficulties.
A bone age study revealed a delay of 2 years (–3 SD). Results of the cardiological examination, brain MRI, and EEG were normal. Further testing was performed, and whole-genome sequencing revealed heterozygous presence of a de novo frameshift mutation in exon 5 of the PPM1D gene (hg19; chr 17: 58734146; NM_003620.3; c.1206_1207del, p.Asn402Lysfs*31). The mutation, which was subsequently confirmed by Sanger sequencing, was not listed on any genetic database and had not previously been reported in the literature.
The most frequent physical characteristics of JDVS include hypotonia,2 delayed bone age,3 feeding difficulties and gastrointestinal problems (including vomiting and constipation),1–4 and such dysmorphic features as the above-mentioned subtle facial abnormalities,2,3 microcephalus, hyperlordosis,4 small hands with brachyphalangia (detectable in 90% of cases), and hypoplastic nails.1–4 The most frequent cognitive and behavioural difficulties are speech delay,4 attention-deficit/hyperactivity disorder,2 and anxiety with an obsessive-compulsive component,2,3 together with hyper- or hyporeactivity to sensory stimuli (hypersensitivity to sound,3 high pain threshold in 90% of cases)2–4; many of these features are present in our patient. Hypersexuality has previously been described in patients with intellectual disability and occasionally in patients with autistic spectrum disorders.5 It is currently unclear whether this hyperactivity is part of a specific behavioural phenotype, which may have not been reported previously.
We report a case of JDVS with characteristic dysmorphic features and no intellectual disability, but presenting surprising hypersexual behaviour, secondary to a novel mutation. Although disability of various degrees is considered the standard in this syndrome,1 the series by Jansen et al.2 includes another case with JDVS and no intellectual disability, highlighting the need for more comprehensive genetic studies in patients with complex neurodevelopmental disorders of unknown origin.
FundingThe authors have received no funding for this study.
Please cite this article as: Martín Fernández-Mayoralas D, Fernández-Perrone AL, Jiménez De Domingo A, Alba Menéndez A, Fernández-Jaén A. Síndrome de Jansen-de Vries. Primer caso diagnosticado en Espa˜na. Neurología. 2021;36:330–332.
