



Early-onset Alzheimer disease (EOAD), which presents in patients younger than 65 years, has frequently been described as having different features from those of late-onset Alzheimer disease (LOAD). This review analyses the most recent studies comparing the clinical presentation and neuropsychological, neuropathological, genetic, and neuroimaging findings of both types in order to determine whether EOAD and LOAD are different entities or distinct forms of the same entity. We observed consistent differences between clinical findings in EOAD and in LOAD.
Fundamentally, the onset of EOAD is more likely to be marked by atypical symptoms, and cognitive assessments point to poorer executive and visuospatial functioning and praxis with less marked memory impairment. Alzheimer-type features will be more dense and widespread in neuropathology studies, with structural and functional neuroimaging showing greater and more diffuse atrophy extending to neocortical areas (especially the precuneus). In conclusion, available evidence suggests that EOAD and LOAD are 2 different forms of a single entity. LOAD is likely to be influenced by ageing-related processes.
La enfermedad de Alzheimer de inicio precoz (EAIP), definida como la que se manifiesta antes de los 65 años de edad, muestra ciertas características diferentes de la enfermedad de Alzheimer de inicio tardío (EAIT). Nuestro objetivo fue analizar los trabajos más actuales que comparan la clínica, la neuropsicología, la patología, la genética y la neuroimagen de la EAIP y la EAIT, para determinar si nos enfrentamos a dos enfermedades distintas o a variantes de una misma entidad. Como resultado, hallamos consistencia en algunas características diferenciales entre los 2 cuadros clínicos. Fundamentalmente, la EAIP comienza con mayor frecuencia con una clínica atípica; la valoración cognitiva muestra mayor afectación de las funciones ejecutiva y visuoespacial y de las praxias, y menor afectación de la memoria; la neuropatología evidencia mayor densidad y una distribución más difusa de la patología tipo Alzheimer; los estudios de neuroimagen estructural y funcional muestran una afectación cortical mayor y más difusa, afectando al neocórtex (especialmente el precuneus). En conclusión, las evidencias actuales hacen pensar que la EAIP y la EAIT son variantes clínicas de una misma entidad, que en el caso de la EAIT se ve influida probablemente por factores asociados al envejecimiento.
Alzheimer disease (AD) is the most frequent neurodegenerative disease. It presents clinically as progressive dementia that predominantly affects episodic memory.1 Age is the main risk factor for developing AD, and in fact, prevalence of the disease is higher in older segments of the population. AD is the most frequent cause of dementia and accounts for approximately 60% of the total cases, whether before2 or after the age of 65,3 the age marking the arbitrary limit between early-onset and late-onset dementia.
In 1907, Alois Alzheimer described the disease now bearing his name today in a 51-year-old woman who had developed dementia with predominant language impairment and behavioural changes.4 For years, this syndrome of early-onset dementia without amnesia was thought to define AD and distinguish it from senile dementia, which was characterised by a later onset and symptoms of amnesia and attributed to the ageing process. However, in the 1960s and 1970s, studies showed that the neuropathology underlying early-onset and senile dementia was the same; as such, both clinical variants are produced by the same disease, AD.5,6 From that time on, once the clinical criteria for an AD diagnosis had been established, doctors were more likely to recognise the senile onset form7 since it was far more common, while the other ‘atypical’ early-onset variant was almost forgotten. However, a number of recent studies have underlined the clinical heterogeneity of AD and investigated its underlying causes.
This review aims to present the similarities and differences between early-onset AD (EOAD) and late-onset AD (LOAD) with regard to the clinical features, neuropsychology, neuropathology, genetics, and neuroimaging features described to date. This will serve to clarify if there are indeed 2 different entities or 2 clinical variants of the same disease.
Clinical presentationThe most common initial clinical presentation of AD is episodic memory loss, which is accompanied by progressive impairment of other cognitive domains. Nevertheless, some patients present alterations in other cognitive areas and memory remains relatively well preserved. AD may even present as a ‘focal’ syndrome in which the predominant symptom is apraxia or an impairment of language, visual function, or visuospatial reasoning. This wide array of cases illustrates the heterogeneous clinical presentations of AD, and this results in major challenges and frequent diagnostic errors.8 Some such ‘atypical’ syndromes can easily be mistaken for other entities, such as frontotemporal dementia (FTD), when executive or language dysfunctions are dominant; or corticobasal degeneration, when there are signs of corticobasal syndrome. Assigning the correct diagnosis to ‘focal’ syndromes of AD (especially when ruling out FTD in the differential diagnosis) will involve detecting any temporal lobe and posterior hemisphere symptoms (amnesia, visuospatial dysfunction) on the one hand, and on the other, verifying whether the focal deficits in these syndromes seem to be less selective (and profound) than those in FTD. Here, an exhaustive neurological examination will reveal deficits in other cognitive domains and those affecting multiple functional systems within a single domain (for example, phonology, spelling, and syntax within language).9
In light of this heterogeneous array, some authors have attempted to facilitate diagnosis by elaborating classification systems for specific AD subtypes: the typical form (memory loss with other deficits) and the temporal variant (with isolated memory loss) are late-onset syndromes. In contrast, the left/language variant (a nonfluent aphasia), progressive logopenic aphasia (with preserved fluency and predominant repetition deficit), right/visuoperceptive variant (including posterior cortical atrophy, which is almost always due to AD) and the frontal/executive variant are early-onset syndromes.10 Using a similar approach, another study grouped AD patients in 3 categories: those with frontal lobe dysfunction, very early onset, and a family history; another with primarily posterior deficit (temporoparietal and/or occipital lobes) and early onset, and a third group with predominantly temporal lobe dysfunction in elderly patients.11 Lastly, current diagnostic criteria for AD describe a typical amnestic presentation and other non-amnestic presentations, including those with predominant deficits in language, visuospatial function, and executive functions.12
Generally speaking, studies coincide in that non-amnestic or atypical clinical presentations are more frequently displayed by patients with EOAD.8,10,13,14 In fact, a third of the patients with EOAD display an atypical presentation, compared to 6% of those with LOAD.8,11 On the other hand, most studies15–18 have concluded that EOAD runs a more aggressive course.
NeuropsychologyNumerous articles have compared the neuropsychological profiles of patients with AD to determine whether some cognitive domains are more affected than others given different ages of onset. Most such studies19–27 indicate significant differences. Here, we offer a summary of the results for each cognitive domain.
MemoryIn general, studies show that the patients with the greatest impairment of this domain have LOAD.16,17,20 Furthermore, memory seems to be relatively well-preserved in the early stages of EOAD with respect to LOAD.19 Specifically, researchers have observed that LOAD has a greater impact on memory, whether of recent events17,20 or of well-consolidated information,16 although some cases only exhibit impaired recognition.21 Other studies22,23 indicate a qualitative difference in impairment with memory loss predominating in EOAD vs memory encoding failure in LOAD.23 Researchers have also observed poorer temporal orientation in the group with LOAD,17,22 which can be attributed to the accentuated memory loss in this group of patients.22
LanguagePatients with LOAD perform more poorly on visual confrontation naming tests, such as the Boston Naming Test.20,21 Some studies reached the same conclusion, but also found that the naming function deteriorated more quickly in EOAD.24 On the other hand, subjects with EOAD score lower on writing tasks.20 Nevertheless, other studies have not found any differences in language,19,25 meaning that the language domain is one of the most controversial when comparing these 2 variants.
Executive functionDifferent studies indicate that patients with EOAD will perform more poorly on complex attention and working memory tasks16,19–21,23 and show more errors on response inhibition tasks.21
Visuospatial functionIn general, patients with EOAD will have poorer results than the late-onset group on visual-cognitive tasks,19,20,26 referring to both the object perception domain19 and the spatial perception19,26 and construction domains.20,26
Learned motor behavioursNumerous studies have shown that patients with EOAD show more severely affected motor behaviours.17,22
Behaviour disturbancesOne study compared the prevalence of psychiatric and behavioural symptoms between subjects with EOAD and LOAD and comparable severity of dementia; it concluded that the late-onset group was more affected. In any case, the fact that dysphoria and apathy are relatively frequent in the EOAD group may be relevant.28
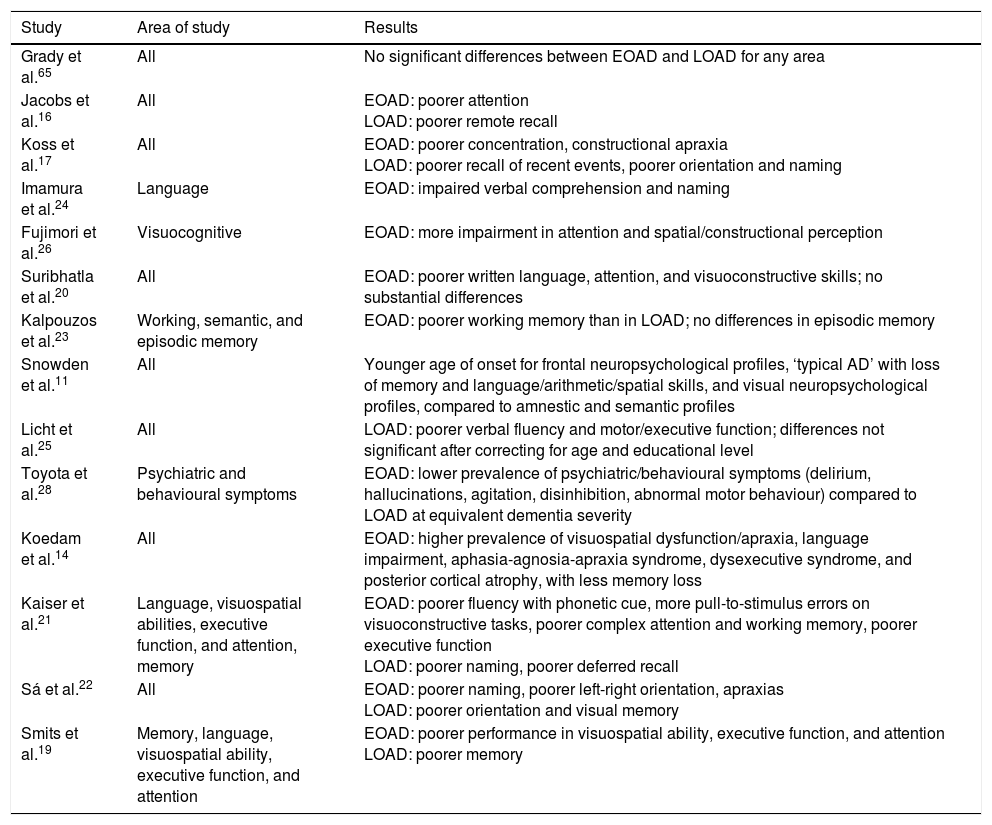
In summary, patients with EOAD perform more poorly on tasks related to written language, executive function, attention, visuospatial abilities, and motor skills, whereas patients with LOAD show poorer performance on tasks involving episodic memory and visual confrontation naming (Table 1).
Summary of the articles consulted on the subject of neuropsychology in EOAD and LOAD.
| Study | Area of study | Results |
|---|---|---|
| Grady et al.65 | All | No significant differences between EOAD and LOAD for any area |
| Jacobs et al.16 | All | EOAD: poorer attention LOAD: poorer remote recall |
| Koss et al.17 | All | EOAD: poorer concentration, constructional apraxia LOAD: poorer recall of recent events, poorer orientation and naming |
| Imamura et al.24 | Language | EOAD: impaired verbal comprehension and naming |
| Fujimori et al.26 | Visuocognitive | EOAD: more impairment in attention and spatial/constructional perception |
| Suribhatla et al.20 | All | EOAD: poorer written language, attention, and visuoconstructive skills; no substantial differences |
| Kalpouzos et al.23 | Working, semantic, and episodic memory | EOAD: poorer working memory than in LOAD; no differences in episodic memory |
| Snowden et al.11 | All | Younger age of onset for frontal neuropsychological profiles, ‘typical AD’ with loss of memory and language/arithmetic/spatial skills, and visual neuropsychological profiles, compared to amnestic and semantic profiles |
| Licht et al.25 | All | LOAD: poorer verbal fluency and motor/executive function; differences not significant after correcting for age and educational level |
| Toyota et al.28 | Psychiatric and behavioural symptoms | EOAD: lower prevalence of psychiatric/behavioural symptoms (delirium, hallucinations, agitation, disinhibition, abnormal motor behaviour) compared to LOAD at equivalent dementia severity |
| Koedam et al.14 | All | EOAD: higher prevalence of visuospatial dysfunction/apraxia, language impairment, aphasia-agnosia-apraxia syndrome, dysexecutive syndrome, and posterior cortical atrophy, with less memory loss |
| Kaiser et al.21 | Language, visuospatial abilities, executive function, and attention, memory | EOAD: poorer fluency with phonetic cue, more pull-to-stimulus errors on visuoconstructive tasks, poorer complex attention and working memory, poorer executive function LOAD: poorer naming, poorer deferred recall |
| Sá et al.22 | All | EOAD: poorer naming, poorer left-right orientation, apraxias LOAD: poorer orientation and visual memory |
| Smits et al.19 | Memory, language, visuospatial ability, executive function, and attention | EOAD: poorer performance in visuospatial ability, executive function, and attention LOAD: poorer memory |
Characteristic neuropathological findings in AD are extracellular deposits of senile plaques consisting of amyloid beta and intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein. Deposition of amyloid plaques begins in the basal regions of the frontal, temporal, and occipital lobes and progresses to affect the primary sensory areas. Neurofibrillary tangles, on the other hand, will first affect the transentorhinal region and progress toward the limbic system before finally reaching the neocortex.29
The brain's distribution of AD-specific pathology is correlated with the type of clinical presentation. In fact, Murray et al.30 describe 3 patterns of neurofibrillary tangle distribution in AD: the typical pattern described above, another pattern in which the hippocampus is spared (with more tangles in the cortex than in the hippocampus and with less hippocampal atrophy), and a predominantly limbic pattern. In our study, curiously enough, senile plaque density was similar in all 3 of the listed patterns. The pattern preserving the hippocampus was associated with early onset, a more aggressive course, and a higher prevalence of atypical presentations (up to 30%). Based on these observations, some researchers have postulated that a slightly different pathological cascade may be at work in patients with an atypical presentation. Here, although onset would also be determined by amyloid deposits, the formation of neurofibrillary tangles occurs earlier and its topographical pattern is very different.27
Various teams have attempted to quantify pathological features in AD to compare patients with early- and late-onset forms of the disease. Generally speaking, authors have determined that typical EOAD patients display a greater density of senile plaques15 and neurofibrillary tangles10,31,32 and greater neuronal loss compared to their counterparts with LOAD14 or to those with atypical EOAD.31 The latter also show more marked impairment of the neocortex than do typical cases.31 This seems to indicate that EOAD has a more aggressive course, but some interpret these findings within the context of the brain's functional reserve; they state that patients with LOAD will not require such a high pathological load as patients with EOAD in order to exhibit clinical symptoms.32
The role of soluble oligomeric forms of amyloid beta in EOAD and LOAD has also been studied; these forms may have a more direct effect on the loss of neural function than does fibrillary amyloid beta. In fact, studies point to a good correlation between the level of oligomers and neurotransmitter activity (measured by choline acetyltransferase).33 They also reveal a distinct pattern of oligomer subtypes for each group: there is a higher level of pentamers in the insoluble fraction in EOAD than in LOAD,33 which indicates potential differences in the pathogenesis of each type.
Lastly, when linking neuropathology findings with clinical expression for these 2 patient groups, we cannot overlook the effect of concomitant illness. In younger patients, the correlation between the amyloid load and the level of dementia is strong, but this is not the case in older patients in whom vascular disease may play a stronger role in dementia.34
In summary, studies indicate that the early-onset form of AD is more extensive, and that differences are more pronounced outside of the temporal lobe.35
Cerebrospinal fluid (CSF) markersThe biomarkers recently developed for CSF are helpful in diagnosing AD, and researchers have examined the possibility that they may be especially useful for EOAD given its atypical presentation. Studies show that levels of amyloid beta, total tau protein, and phosphorylated tau protein are abnormal in both EOAD and LOAD, but values are similar for both groups.36,37 On the other hand, there seems to be a correlation between the (pathologically low) level of amyloid beta in CSF and the degree of brain atrophy in areas that are specific to each variant (especially the precuneus in EOAD and the hippocampus in LOAD).37 Among EOAD cases, there does seem to be a difference between typical and atypical profiles; the latter display a higher level of total tau in CSF, regardless of disease duration and the degree of clinical severity. This finding indicates more intense degeneration in patients with an atypical profile.38
GeneticsIt cannot be overlooked that AD may be caused by an autosomal dominant mutation on the PSEN1, PSEN2, or APP gene.35 These cases account for less than 1% of all patients with AD and they are usually characterised by early onset35; nevertheless, they make up only a small percentage of all patients with EOAD and remain outside of the scope of this article.
Allele ¿4 of apolipoprotein E (APOE*4) is the most important risk factor for sporadic AD. Presence of this allele, whether heterozygous or homozygous, has been associated with a younger age of onset of the disease.35,39,40 However, various neuroimaging studies coincide in that they show more pronounced atrophy of the medial temporal lobe and especially the hippocampus in patients carrying the ¿4 allele in APOE compared to non-carriers, regardless of age of onset of the disease.41,42 There was more extreme hypometabolism in the medial temporal lobe in ¿4/¿4 patients than in those with the ¿3/¿3 genotype within the EOAD group, but no genotype-related differences in the LOAD group.43 This is consistent with the greater prevalence of bearers of this allele among patients with amnestic presentation9,35 and the lower percentage of bearers among patients exhibiting preservation of the hippocampus.29 To summarise, we may therefore state that although the ¿4 allele is linked to younger age at AD onset, this only occurs in patients with typical AD who necessarily belong to the LOAD group, affecting the younger patients in that group.35
On the other hand, some studies have indicated that APOE*4 determines EOAD progression.17,35 As such, in the absence of ¿4, progression is quicker for EOAD than for LOAD; where ¿4 is present, progression is similar in the 2 groups.35 Likewise, patients with EOAD and the ¿4 allele have been found to be at greater risk of developing myoclonias with less tremor; this indicates a different clinical course, determined by the APOE genotype, within the group of patients with EOAD.35
It should be understood that, in recent years, genome-wide association studies (GWAS) have detected polymorphisms in certain genes associated with LOAD, such as CLU, CR1, PICALM, SORL1, BIN1, CTNNA3, GAB2, DNMBP, ABCA7, TREM2, and TOMM40; the latter is associated with an earlier onset of LOAD determined by the APOE genotype.44
Structural neuroimagingStructural neuroimaging studies, including computed tomography (CT) and magnetic resonance (MR) studies, are extremely important for the assessment of patients with cognitive impairment, and they are mainly used to rule out secondary causes. Furthermore, MR has shown high sensitivity as a means of evaluating the cerebral atrophy associated with neurodegenerative processes; in AD, atrophy typically affects the medial temporal region and extends posteriorly to the parietotemporal and frontal regions.45
Assessment of overall cerebral volumeMultiple MR studies have calculated the rate of cerebral atrophy in patients with AD and found it to be greater in EOAD, with a decrease of 2% to 3% of total brain volume in a year46 vs 0.8% in LOAD patients47 (note that the first study included both familial and sporadic cases). The rate of atrophy for all patients in general is 1.4% and 0.6% in healthy controls.48 Furthermore, studies indicate that the atrophy rate increases by 0.32% per year2 in patients with EOAD46; on the other hand, studies of AD making no distinctions by age of onset have not pointed to significant accelerations in atrophy and indicate a mean value of 0.09% per year2.48 All of the above describes EOAD as having a more aggressive course.
Assessment of grey matterNumerous studies have compared atrophy patterns in grey matter between patients with EOAD and LOAD. Frisoni et al.49 found more pronounced overall atrophy in EOAD than in the late-onset form (19.5% vs 11.9%). Atrophy primarily affected neocortical areas, especially the occipital lobe in EOAD and the hippocampus in LOAD, and these topographies correspond with the 2 different clinical patterns. Later studies have replicated these findings and attached special relevance to the precuneal atrophy occurring in EOAD.50–52 Furthermore, the cortical maps that Frisoni et al.49 compared showed that atrophy was very diffuse in EOAD, whereas in LOAD it was directed at the temporal lobe and the temporoparietal junction.53
We know of only one longitudinal study carried out to date that has compared progression of cortical thinning between these groups; it revealed, for EOAD, more rapid and diffuse atrophy of the association cortices (the left middle and frontal gyri, left inferior parietal lobe, posterior area of the left superior temporal gyrus, left fusiform gyrus, bilateral posterior cingular gyri, and precuneus) and comparatively more atrophy of the left parahippocampal gyrus in LOAD.54 This pattern of atrophy corresponded to the more pronounced clinical impairment displayed by the EOAD group in the areas of executive functions, attention, and language.54
One recent study investigated impairment in the deep cerebral nuclei in EOAD and LOAD.55 The different patient groups display distinct topographical atrophy patterns in the striate. Patients with EOAD exhibited more extensive atrophy of the dorsal striatum, compared to the ventral striatum in those with LOAD. The authors’ interpretation was that the changes might have been secondary to loss of afferences of the medial temporal lobe in the case of the ventral striatum, and of the parietal lobe for the dorsal striatum. Another longitudinal study compared the atrophy rate of subcortical structures between these 2 patient groups.56 It found greater losses of volume in the caudate, putamen, and thalamus in patients with EOAD than in those with LOAD, whereas losses in the hippocampus and amygdala followed similar patterns. These findings may be related to the fact that frontal functions decline more rapidly than memory does in EOAD.
Assessment of white matterStudies have pointed to a predominantly parahippocampal pattern of atrophy in LOAD, compared to a more diffuse pattern of posterior atrophy in EOAD that primarily affects the splenium of the corpus callosum and the dorsal temporoparietal regions. These white-matter findings coincide with the topography of affected grey matter, indicating that white matter atrophy is secondary to atrophy of the grey matter.41
A recent study used diffusion tensor imaging (DTI) to compare white matter microstructural damage between the 2 patient groups.57 These authors observed more severe and diffuse atrophy in patients with EOAD, and they describe damage to the interhemispheric connections, limbic system and main associative pathways, and the posterior cingulate cortex; LOAD was observed to affect mainly the corpus callosum.
In summary, EOAD displays a more diffuse atrophy pattern in both grey and white matter, and it affects neocortical areas (especially the precuneus); atrophy in LOAD is restricted to the hippocampus. Furthermore, progression seems to be quicker in EOAD, and it extends to subcortical areas, mostly to the putamen.
Functional neuroimagingFunctional neuroimaging techniques allow doctors to assess numerous functions of the brain. One of the most widely-used techniques is positron emission tomography (PET) with 18F-fluorodeoxyglucose (FDG), which estimates neural activity based on the regional rate of glucose uptake.45 Single photon emission computed tomography (SPECT) with 99mTc-HMPAO (99mTc-hexamethylpropyleneamineoxime) estimates neural activity by providing an image of regional blood flow in the brain.45 On the other hand, functional MRI lets us analyse the brain's response to specific stimuli, which is reflected by the haemodynamic changes in the brain regions activated by each stimulus.
PET of glucose metabolismThe typical pattern of AD anomalies in PET studies consists of hypometabolism in the posterior cingulate, the posterior temporoparietal cortex, and the anterior region of the medial temporal lobes.45 Hypometabolism in these regions is more marked in EOAD than in LOAD according to studies employing region of interest analysis techniques (examining structures that are chosen a priori), and also according to voxel-based techniques (analysing the brain as a whole with no need for a priori selection of specific regions).45,58,59 Given the same degree of functional impairment according to the Clinical Dementia Rating (CDR), the patients with EOAD showed greater hypometabolism, but this could be attributed to the younger subjects’ greater functional reserve. Furthermore, the hypometabolism curve is more pronounced than that of the CDR, which indicates that the metabolic dysfunction progresses more rapidly.59
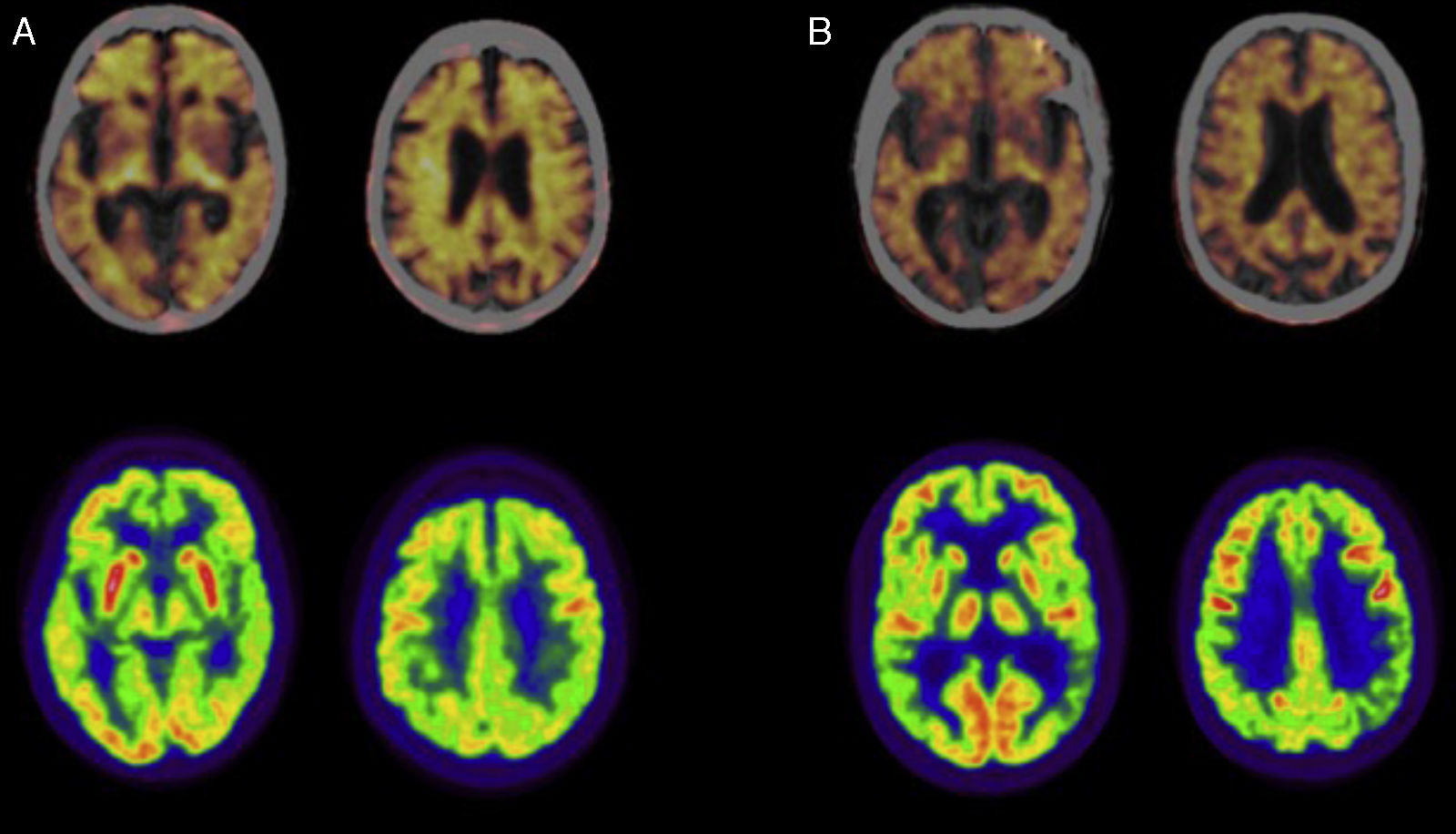
Fig. 1 shows a PET-FDG study from a patient with LOAD and another from a patient with EOAD (lower row).
 studies with florbetapir (upper row; images fused with CT) and 18F-fluorodeoxyglucose (FDG; lower row) in patients diagnosed with Alzheimer disease. (A) Woman aged 79 diagnosed with typical Alzheimer disease. The PET study with florbetapir gives a positive result for cortical amyloid plaques. The PET-FDG study indicates hypometabolism in the posterior association cortex, predominantly on the left side. (B) Man aged 57 diagnosed with left/language variant Alzheimer disease. The PET study with florbetapir gives a positive result for cortical amyloid plaques. The PET-FDG study indicates hypometabolism in the posterior association cortex, predominantly on the left side.")
Positron emission tomography (PET) studies with florbetapir (upper row; images fused with CT) and 18F-fluorodeoxyglucose (FDG; lower row) in patients diagnosed with Alzheimer disease. (A) Woman aged 79 diagnosed with typical Alzheimer disease. The PET study with florbetapir gives a positive result for cortical amyloid plaques. The PET-FDG study indicates hypometabolism in the posterior association cortex, predominantly on the left side. (B) Man aged 57 diagnosed with left/language variant Alzheimer disease. The PET study with florbetapir gives a positive result for cortical amyloid plaques. The PET-FDG study indicates hypometabolism in the posterior association cortex, predominantly on the left side.
The pattern normally seen using SPECT in typical AD is similar to that described using PET.45 Furthermore, patients with EOAD exhibit marked hypoperfusion of the posterior associative cortical areas, whereas late-onset cases will show hypoperfusion in the medial temporal areas.60
In summary, evidence points once again to more pronounced impairment of the neocortex (parieto-occipital and even frontal) in patients with EOAD, whereas atrophy in LOAD is more limited to the medial temporal lobe.
Brain functional magnetic resonance imagingFunctional MRI was used in one study that attempted to characterise functional connectivity patterns in EOAD and LOAD.61 The EOAD group displayed decreased connectivity of the dorsolateral prefrontal network (DLPFN, related to executive function) and increased connectivity in the anterior temporal network (ATN, related to declarative memory); patients with LOAD exhibited the opposite pattern. The authors linked their findings to the distinct topographies of neural damage (decreased connectivity) in each of the 2 groups, and the fact that damage would give rise to compensatory mechanisms (increased connectivity).
PETThe recent development of radiotracers able to bind to amyloid protein has made it possible to evaluate AD features in vivo.45 Most of the studies performed using Pittsburgh compound B (PiB) in patients with EOAD and LOAD did not reveal significant intergroup differences.55,62 According to the authors, the discrepancy between PET-FDG images (which differ significantly between groups, as mentioned) and PET-PiB images (which are similar in both groups) indicates that other pathological processes, such as the formation of neurofibrillary tangles, neuroinflammation, or amyloid oligomers (mentioned previously and not visible with PET-PiB), may be more involved than fibrillar amyloid deposition in eliciting metabolic changes.62 Nevertheless, Ossenkoppele et al.63 observed a greater concentration of the radiotracer in the parietal cortex of patients with EOAD and a recent study showed that patients with EOAD displayed more amyloid deposition in the basal ganglia, thalamus, left superior temporal cortex, and left cuneus.64 This is to be expected based on the PET-FDG metabolic neuroimaging studies that we have already described, and it would fit the idea of a pathological cascade in which deposition of amyloid beta would precede metabolic dysfunction. It remains to be seen what would cause amyloid deposits in certain areas and not in others.63
Fig. 1 also shows a PET study of amyloid deposits in one patient with LOAD and another with EOAD (upper row).
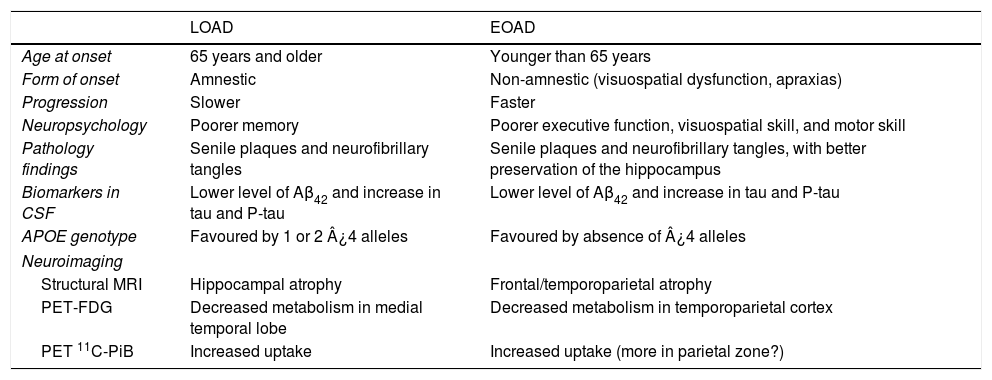
ConclusionEOAD and LOAD have many traits in common, but they also exhibit numerous differences (Table 2). The fact that they display the same pathological process, which is the only defining diagnostic criterion for AD, obliges us to conclude that they are technically variants of the same disease. Nevertheless, the substantial differences between these variants, which we have listed in this study, raise the question of whether anatomical pathology findings alone should constitute what defines a nosological entity when it comes to classifying these neurodegenerative diseases.
General characteristics of EOAD and LOAD.
| LOAD | EOAD | |
|---|---|---|
| Age at onset | 65 years and older | Younger than 65 years |
| Form of onset | Amnestic | Non-amnestic (visuospatial dysfunction, apraxias) |
| Progression | Slower | Faster |
| Neuropsychology | Poorer memory | Poorer executive function, visuospatial skill, and motor skill |
| Pathology findings | Senile plaques and neurofibrillary tangles | Senile plaques and neurofibrillary tangles, with better preservation of the hippocampus |
| Biomarkers in CSF | Lower level of Aβ42 and increase in tau and P-tau | Lower level of Aβ42 and increase in tau and P-tau |
| APOE genotype | Favoured by 1 or 2 ¿4 alleles | Favoured by absence of ¿4 alleles |
| Neuroimaging | ||
| Structural MRI | Hippocampal atrophy | Frontal/temporoparietal atrophy |
| PET-FDG | Decreased metabolism in medial temporal lobe | Decreased metabolism in temporoparietal cortex |
| PET 11C-PiB | Increased uptake | Increased uptake (more in parietal zone?) |
In addition, neuropathology and neuroimaging findings pose interesting questions regarding whether or not the underlying pathophysiological process is the same in both forms. Perhaps the differences between these clinical variants can only be explained by the factors involved in ageing. On the other hand, the seemingly more aggressive course of EOAD calls for further investigation of why the disease would exhibit different behaviour in some cases and not in others.
In summary, EOAD more frequently exhibits an atypical early clinical course; furthermore, patients will be more affected in the areas of executive function, visuospatial function, and motor skills, and display less memory loss. EOAD cases show a greater density of amyloid plaques and a more diffuse distribution pattern than that seen in LOAD. Lastly, EOAD cases will show a greater extension of atrophy with a more diffuse pattern and more rapid progression; these tendencies are also observed using PET and SPECT functional imaging. On the other hand, the least consistent results are provided by PET studies evaluating the brain's amyloid load.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Tellechea P, Pujol N, Esteve-Belloch P, Echeveste B, García-Eulate MR, Arbizu J, et al. Enfermedad de Alzheimer de inicio precoz y de inicio tardío: ¿son la misma entidad? Neurología. 2018;33:244–253.
