



La enfermedad de Alzheimer de inicio precoz (EAIP), definida como la que se manifiesta antes de los 65 años de edad, muestra ciertas características diferentes de la enfermedad de Alzheimer de inicio tardío (EAIT). Nuestro objetivo fue analizar los trabajos más actuales que comparan la clínica, la neuropsicología, la patología, la genética y la neuroimagen de la EAIP y la EAIT, para determinar si nos enfrentamos a dos enfermedades distintas o a variantes de una misma entidad. Como resultado, hallamos consistencia en algunas características diferenciales entre los 2 cuadros clínicos. Fundamentalmente, la EAIP comienza con mayor frecuencia con una clínica atípica; la valoración cognitiva muestra mayor afectación de las funciones ejecutiva y visuoespacial y de las praxias, y menor afectación de la memoria; la neuropatología evidencia mayor densidad y una distribución más difusa de la patología tipo Alzheimer; los estudios de neuroimagen estructural y funcional muestran una afectación cortical mayor y más difusa, afectando al neocórtex (especialmente el precuneus). En conclusión, las evidencias actuales hacen pensar que la EAIP y la EAIT son variantes clínicas de una misma entidad, que en el caso de la EAIT se ve influida probablemente por factores asociados al envejecimiento.
Early-onset Alzheimer disease (EOAD), which presents in patients younger than 65 years, has frequently been described as having different features from those of late-onset Alzheimer disease (LOAD). This review analyses the most recent studies comparing the clinical presentation and neuropsychological, neuropathological, genetic, and neuroimaging findings of both types in order to determine whether EOAD and LOAD are different entities or distinct forms of the same entity. We observed consistent differences between clinical findings in EOAD and in LOAD.
Fundamentally, the onset of EOAD is more likely to be marked by atypical symptoms, and cognitive assessments point to poorer executive and visuospatial functioning and praxis with less marked memory impairment. Alzheimer-type features will be more dense and widespread in neuropathology studies, with structural and functional neuroimaging showing greater and more diffuse atrophy extending to neocortical areas (especially the precuneus). In conclusion, available evidence suggests that EOAD and LOAD are 2 different forms of a single entity. LOAD is likely to be influenced by ageing-related processes.
La enfermedad de Alzheimer (EA) es la enfermedad neurodegenerativa más frecuente y se presenta clínicamente como una demencia progresiva con afectación predominante de la memoria episódica1. La edad es el principal factor de riesgo para desarrollar EA y, de hecho, la prevalencia de esta entidad aumenta con el envejecimiento. La EA es la causa más frecuente de demencia, acaparando alrededor de un 60% de los casos, tanto antes2 como después de los 65 años3, edad en la que se establece el límite arbitrario entre demencia de inicio precoz y tardío.
En 1907, Alois Alzheimer describió la enfermedad que hoy lleva su nombre en una paciente de 51 años que desarrolló una demencia con afectación predominante del lenguaje y cambios conductuales4. Durante años, este cuadro de demencia de inicio precoz y clínica no amnésica se consideró el propio de la EA, diferenciándolo de la demencia senil, de inicio más tardío, clínica predominantemente amnésica y atribuida al propio envejecimiento. Sin embargo, en los años 60 y 70 del siglo xx se demostró que la neuropatología era la misma en la demencia de inicio precoz y en la senil, y por tanto, ambas formas clínicas se debían a una sola enfermedad, la EA5,6. A partir de entonces, al establecerse los criterios clínicos de la EA, se tuvo en cuenta el cuadro propio de la forma «senil»7 por ser mucho más habitual y fue cayendo en el olvido la existencia de ese otro subtipo «atípico» de inicio precoz; hasta que recientemente numerosas investigaciones han retomado la descripción de la heterogeneidad clínica de la EA, así como la búsqueda de las causas que la subyacen.
El objetivo de esta revisión es exponer las similitudes y diferencias entre la EA de inicio precoz (EAIP) y la EA de inicio tardío (EAIT) en cuanto a la clínica, la neuropsicología, la neuropatología, la genética y la neuroimagen descritas hasta el momento actual, para poder aclarar si nos enfrentamos a 2 entidades diferentes o a variantes clínicas de la misma enfermedad.
ClínicaLa presentación clínica inicial más común de la EA es el déficit de memoria episódica, que se acompaña de la afectación progresiva de otros dominios cognitivos. Sin embargo, algunos pacientes presentan alteraciones de otras áreas cognitivas, con una relativa preservación de la memoria. De hecho, la EA puede presentarse como síndromes «focales», en que el síntoma predominante sea un problema del lenguaje, de la función visual o visuoespacial o una apraxia. Todo este conjunto de casos contribuye a una realidad clínica heterogénea dentro de la EA, que implica verdaderos retos y frecuentes errores diagnósticos8. En este sentido, algunos de estos síndromes «atípicos» pueden confundirse fácilmente con otras entidades, como la demencia frontotemporal (DFT), cuando predomina la disfunción ejecutiva o del lenguaje, o la degeneración corticobasal, cuando se evidencia un síndrome corticobasal. Se ha indicado que para el correcto diagnóstico de síndromes «focales» de EA como tales (sobre todo en el diagnóstico diferencial con la DFT) conviene, por un lado, fijarse en la existencia de síntomas de lóbulo temporal y hemisferio posterior (amnesia, disfunción visuoespacial) y, por otro lado, en que los déficits focales de estos síndromes parecen ser menos selectivos (y profundos) que los de la DFT, revelando en un examen neurológico exhaustivo fallos de otros dominios cognitivos, y afectando a varios sistemas funcionales dentro de un dominio (p. ej., dentro del lenguaje, a fonología, ortografía, sintaxis…)9.
Algunos autores, a la vista de esta heterogeneidad y con intención de facilitar el diagnóstico, han elaborado clasificaciones de subtipos concretos de la EA: el típico (afectación de la memoria junto con otros déficits) y la variante temporal (con un déficit mnésico aislado) serían síndromes de inicio tardío, mientras que la variante izquierda o de lenguaje (una afasia no fluente), la afasia logopénica progresiva (con fluencia conservada y una afectación predominante en la repetición), la variante derecha o visuoperceptiva (incluyendo la atrofia cortical posterior, casi siempre atribuible a la EA) y la variante frontal o ejecutiva presentarían un inicio precoz10. En la misma línea, otro estudio agrupa a los pacientes con EA en 3 grupos: uno con disfunción frontal, inicio muy precoz e historia familiar, otro con afectación predominantemente posterior (de los lóbulos temporoparietal y/u occipital) e inicio temprano, y un grupo con disfunción predominante del lóbulo temporal en pacientes con una edad más avanzada11. Por último, en los criterios diagnósticos actuales de demencia por EA se establece una presentación amnésica típica y otras presentaciones no amnésicas destacando la afectación predominante del lenguaje, la función visuoespacial o la función ejecutiva12.
Así pues, en general, los estudios coinciden en que las presentaciones clínicas no amnésicas (o atípicas) son más frecuentes en los pacientes con EAIP8,10,13,14. De hecho, un tercio de los pacientes con EAIP tienen una presentación atípica respecto al 6% de pacientes con EAIT8,11. Por otro lado, la mayoría de los estudios15-18 coinciden en atribuir a la EAIP un curso más agresivo.
NeuropsicologíaExisten numerosos trabajos que han comparado el perfil neuropsicológico de los pacientes con EA para establecer si existen dominios cognitivos más afectados en los pacientes según la edad de inicio de la enfermedad. La mayoría de estos estudios han evidenciado19-27 diferencias significativas. Conviene que abordemos los resultados por cada una de las áreas cognitivas.
MemoriaEn general, los estudios muestran una mayor afectación de este dominio en los pacientes con EAIT16,17,20. Más aún, la memoria parece estar relativamente preservada en estadios precoces en los individuos con EAIP en comparación con la EAIT19. Concretamente, se ha observado una mayor afectación del recuerdo, tanto de eventos recientes17,20 como de información bien aprendida16 en este último grupo de pacientes, aunque en algunos casos solo encuentran una afectación del reconocimiento21. Otros estudios22,23 indican una diferente afectación cualitativa, predominando un fallo del recuerdo en la EAIP, y un fallo de la codificación en la EAIT23. También se observa una peor orientación temporal en el grupo con EAIT17,22, atribuible a la mayor pérdida de memoria en este grupo de pacientes22.
LenguajeLos pacientes con EAIT tienen un peor rendimiento en pruebas de denominación por confrontación visual, como en el Boston Naming Test20,21. Algunos estudios encuentran que, aunque lo anterior sea cierto, la función de denominación se deteriora de manera más rápida en la EAIP24. Por otro lado, los sujetos con EAIP obtienen rendimientos inferiores en escritura20. De todos modos, hay estudios que no encuentran diferencias en el lenguaje19,25, lo que deja al área del lenguaje como una de las más controvertidas en el estudio comparativo de estos 2 cuadros.
Función ejecutivaDistintos trabajos indican que los pacientes de EAIP muestran un peor rendimiento en atención compleja y memoria de trabajo16,19-21,23, así como más fallos en tareas de inhibición de respuesta21.
Función visuoespacialLos pacientes con EAIP muestran, en general, peores resultados que los de inicio tardío en pruebas visuocognitivas19,20,26, tanto en el dominio de percepción de objetos19, como en el de percepción espacial19,26 y en el de construcción20,26.
PraxiasVarios estudios han mostrado que los pacientes con EAIP presentan una mayor afectación de las praxias17,22.
Síntomas conductualesUn estudio ha comparado la prevalencia de síntomas psiquiátrico-conductuales entre sujetos con EAIP y EAIT partiendo de una severidad de demencia comparable, observando una mayor afectación en el grupo de inicio tardío. De todos modos, puede ser de interés el hecho de que la disforia y la apatía ocurran con relativa alta frecuencia en el grupo de EAIP28.
En resumen, los pacientes con EAIP puntúan peor en lenguaje escrito, función ejecutiva, atención, habilidades visuoespaciales y praxias, mientras que los pacientes con EAIT presentan un peor rendimiento en memoria episódica y denominación por confrontación visual (tabla 1).
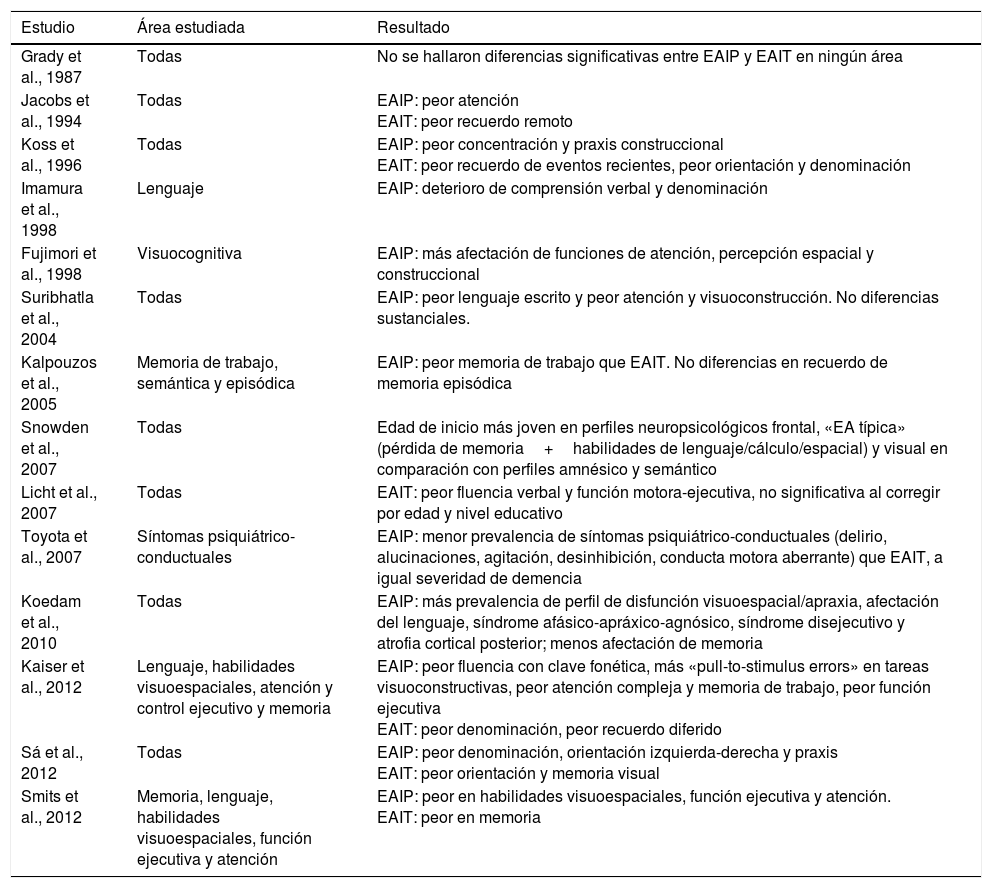
Tabla resumen de los artículos revisados acerca de la neuropsicología de la EAIP y la EAIT
| Estudio | Área estudiada | Resultado |
|---|---|---|
| Grady et al., 1987 | Todas | No se hallaron diferencias significativas entre EAIP y EAIT en ningún área |
| Jacobs et al., 1994 | Todas | EAIP: peor atención EAIT: peor recuerdo remoto |
| Koss et al., 1996 | Todas | EAIP: peor concentración y praxis construccional EAIT: peor recuerdo de eventos recientes, peor orientación y denominación |
| Imamura et al., 1998 | Lenguaje | EAIP: deterioro de comprensión verbal y denominación |
| Fujimori et al., 1998 | Visuocognitiva | EAIP: más afectación de funciones de atención, percepción espacial y construccional |
| Suribhatla et al., 2004 | Todas | EAIP: peor lenguaje escrito y peor atención y visuoconstrucción. No diferencias sustanciales. |
| Kalpouzos et al., 2005 | Memoria de trabajo, semántica y episódica | EAIP: peor memoria de trabajo que EAIT. No diferencias en recuerdo de memoria episódica |
| Snowden et al., 2007 | Todas | Edad de inicio más joven en perfiles neuropsicológicos frontal, «EA típica» (pérdida de memoria+habilidades de lenguaje/cálculo/espacial) y visual en comparación con perfiles amnésico y semántico |
| Licht et al., 2007 | Todas | EAIT: peor fluencia verbal y función motora-ejecutiva, no significativa al corregir por edad y nivel educativo |
| Toyota et al., 2007 | Síntomas psiquiátrico-conductuales | EAIP: menor prevalencia de síntomas psiquiátrico-conductuales (delirio, alucinaciones, agitación, desinhibición, conducta motora aberrante) que EAIT, a igual severidad de demencia |
| Koedam et al., 2010 | Todas | EAIP: más prevalencia de perfil de disfunción visuoespacial/apraxia, afectación del lenguaje, síndrome afásico-apráxico-agnósico, síndrome disejecutivo y atrofia cortical posterior; menos afectación de memoria |
| Kaiser et al., 2012 | Lenguaje, habilidades visuoespaciales, atención y control ejecutivo y memoria | EAIP: peor fluencia con clave fonética, más «pull-to-stimulus errors» en tareas visuoconstructivas, peor atención compleja y memoria de trabajo, peor función ejecutiva EAIT: peor denominación, peor recuerdo diferido |
| Sá et al., 2012 | Todas | EAIP: peor denominación, orientación izquierda-derecha y praxis EAIT: peor orientación y memoria visual |
| Smits et al., 2012 | Memoria, lenguaje, habilidades visuoespaciales, función ejecutiva y atención | EAIP: peor en habilidades visuoespaciales, función ejecutiva y atención. EAIT: peor en memoria |
Los hallazgos neuropatológicos característicos de la EA son el depósito extracelular de placas seniles de β-amiloide y los ovillos neurofibrilares intracelulares, compuestos por proteína tau hiperfosforilada. El depósito de las placas de amiloide se inicia en las porciones basales de los lóbulos frontales, temporales y occipitales, progresando hasta llegar a afectar a las áreas sensoriales primarias. Los ovillos neurofibrilares, por otro lado, comienzan por afectar a la región transentorrinal progresando hacia el sistema límbico y, por último, al neocórtex29.
La distribución en el cerebro de la patología propia de la EA se correlaciona con el tipo de presentación clínica. De hecho, Murray et al.30 describen 3 patrones de distribución de depósito de los ovillos neurofibrilares en la EA: el típico (descrito previamente), el que preserva el hipocampo (con más ovillos en la corteza que en el hipocampo y con una menor atrofia de esta estructura) y el predominantemente límbico. Curiosamente, en este trabajo, la densidad de placas seniles fue similar en los 3 patrones mencionados. El patrón de preservación hipocampal se asocia a un inicio precoz, un curso más agresivo y una mayor prevalencia de presentaciones atípicas (de hasta el 30%). Se ha llegado a postular, a partir de estas observaciones, la existencia de una cascada patológica algo distinta en los pacientes con presentación atípica, en la que, aunque el inicio venga determinado también por el depósito de amiloide, la formación de ovillos neurofibrilares es más precoz y sigue un patrón topográfico muy distinto27.
Varios grupos han querido comparar la cantidad de patología tipo EA en pacientes con inicio precoz y tardío. En general, los autores han encontrado una mayor densidad de placas seniles15 y ovillos neurofibrilares10,31,32, con una mayor pérdida neuronal, en pacientes con EAIP (vs. EAIT)14 o EAIT con clínica atípica (vs. clínica típica)31, teniendo además estos una mayor afectación del neocórtex que los casos típicos31. Esto parece apuntar a una mayor agresividad de la EAIP, pero algunos lo interpretan en el marco de la reserva funcional, afirmando de los pacientes con EAIT no precisan de tanta carga patológica para mostrar síntomas clínicos como los pacientes con inicio más precoz32.
También se ha estudiado el papel de los oligómeros solubles de β-amiloide en EAIP y EAIT, pues se postula que estos tienen un efecto más directo sobre la pérdida de función neuronal que el β-amiloide fibrilar. De hecho, se ha comprobado una buena correlación entre el nivel de oligómeros y la actividad neurotransmisora (medida por actividad de la colinacetiltransferasa)33, y se ha detectado un patrón distinto de subtipos de oligómeros en cada grupo, caracterizado sobre todo por un nivel mayor de pentámeros en la fracción insoluble en EAIP comparado con EAIT33, lo que apunta a posibles diferencias en la patogenia de ambos.
Por último, a la hora de relacionar los hallazgos neuropatológicos con la expresión clínica en estos 2 grupos de pacientes, no podemos olvidar el potencial papel de la patología concomitante. En pacientes jóvenes, la correlación entre la carga patológica y el nivel de demencia es fuerte, mientras que no es así en pacientes de mayor edad, pudiendo esto reflejar un mayor papel de la patología vascular en este grupo de pacientes34.
En resumen, los estudios apuntan a una afectación más extensa de la patología tipo EA en la forma de inicio precoz, siendo las diferencias más notables fuera del lóbulo temporal35.
Marcadores de líquido cefalorraquídeoEn los últimos años se han desarrollado biomarcadores en el líquido cefalorraquídeo (LCR) como ayuda en el diagnóstico de la EA y se ha estudiado la posibilidad de que tengan una utilidad especial en el marco de la EAIP dada su presentación atípica. Se ha hallado que los niveles de β-amiloide, tau total y tau fosforilada son anormales tanto en la EAIP como en la EAIT, pero con unos valores similares en ambos grupos36,37. Por otro lado, parece haber una correlación entre el nivel (patológicamente bajo) de β-amiloide en LCR y el grado de atrofia cerebral en áreas específicas de cada síndrome (sobre todo precúneo en la EAIP e hipocampo en la EAIT)37. Dentro de la EAIP, sí parece haber una diferencia entre los casos con clínica típica y atípica, teniendo estos últimos un nivel más alto de tau total en el LCR, independientemente de la duración de la enfermedad y el índice de severidad clínica. Esto indica una degeneración más intensa en pacientes con clínica atípica38.
GenéticaEs necesario mencionar que la EA puede deberse a una mutación autosómica dominante en los genes PSEN1, PSEN2 o APP35. Estos casos suponen menos de un 1% de los pacientes con EA y suelen caracterizarse por una edad de inicio precoz35; sin embargo, constituyen solo una pequeña proporción de los pacientes con EAIP y no son el objeto de estudio de este trabajo.
El alelo ¿4 de la apolipoproteína E (ApoE4) es el factor de riesgo genético más importante para la EA esporádica. La presencia de este alelo, en heterocigosis u homocigosis, se ha asociado a una reducción en la edad de inicio de la enfermedad35,39,40. Sin embargo, diversos estudios de neuroimagen coinciden en mostrar una mayor atrofia del lóbulo temporal medial y, sobre todo, del hipocampo en pacientes portadores del alelo ¿4 de APOE respecto a los no portadores, independientemente de la edad de inicio de la enfermedad41,42. También se ha hallado un mayor hipometabolismo en el lóbulo temporal medial en pacientes ¿4/¿4 respecto al genotipo ¿3/¿3 en el grupo de la EAIP, no encontrándose diferencias en la EAIT43. Esto es consistente con la mayor prevalencia de portadores de este alelo entre los pacientes con una presentación de tipo amnésico9,35 y de una menor proporción entre los pacientes con un patrón de preservación del hipocampo29. Por tanto, y en resumen, diríamos que, aunque el alelo ¿4 se relaciona con una edad más temprana de presentación, lo hace entre los pacientes con EA típica (pertenecientes, por tanto, más bien al grupo de EAIT, si bien los más jóvenes entre ellos)35.
Por otro lado, algunos estudios han indicado que el alelo ApoE4 condiciona la progresión de la EAIP17,35. De tal forma que, en ausencia del alelo ¿4, la progresión en la EAIP es más rápida que en la EAIT, mientras que, en presencia del alelo, es similar en los 2 grupos35. Así mismo, se ha encontrado entre los pacientes con EAIP con alelo ¿4 un mayor riesgo de desarrollar mioclonías y menos temblor, lo que indica una diferente evolución clínica en función del genotipo ApoE dentro del grupo de pacientes con EAIP35.
Cabe mencionar que, en los últimos años, estudios de asociación de genoma completo (GWAS) han encontrado polimorfismos en algunos genes que se asocian a EAIT, como son CLU, CR1, PICALM, SORL1, BIN1, CTNNA3, GAB2, DNMBP, ABCA7, TREM2 o TOMM40 (asociándose este último a un inicio más precoz dentro de la EAIT según el genotipo de ApoE)44.
Neuroimagen estructuralLa realización de pruebas de neuroimagen estructural, como la tomografía computarizada (TC) o la resonancia magnética (RM), es de gran importancia en el estudio de pacientes con deterioro cognitivo, principalmente para descartar causas secundarias. Además, la RM ha demostrado tener una alta sensibilidad para valorar la atrofia cerebral asociada a los procesos neurodegenerativos, que en el caso de la EA afecta típicamente a la región temporal medial y, posteriormente, a regiones parietotemporales y frontales45.
Valoración del volumen cerebral globalVarios estudios de RM han calculado la tasa de atrofia cerebral en pacientes con EA, hallando que es mayor en la EAIP, con una disminución de 2-3% del volumen cerebral total al año46 (nótese que en este estudio se incluyó a pacientes con EA esporádica, pero también casos con EA familiar), frente a un 0,8% en pacientes de inicio tardío47, un 1,4% en la EA en general y un 0,6% en controles sanos48. Además, se encontró una aceleración progresiva de la atrofia de un 0,32%/año2,46 en el grupo de EAIP, mientras que en estudios con pacientes con EA, sin diferenciar por edad de inicio, no se ha hallado una aceleración significativa de la atrofia (siendo el valor medio 0,09%/año2)48. Todo esto apunta a un curso más agresivo de la EAIP.
Valoración de la sustancia grisVarios estudios han comparado el patrón de atrofia de la sustancia gris en pacientes con EAIP y EAIT. Frisoni et al.49 hallaron una atrofia global mayor en la EAIP frente a la de inicio tardío (19,5% vs. 11,9%). La atrofia correspondía principalmente a áreas neocorticales, sobre todo, el lóbulo occipital en la EAIP y al hipocampo en la EAIT, lo que se corresponde con la diferente afectación clínica. Estudios posteriores han replicado estos hallazgos, dando especial importancia a la atrofia del precúneo en la enfermedad de inicio precoz50-52. Además, en el trabajo de Frisoni et al. la comparación de mapas corticales mostró que la atrofia en la EAIP era muy difusa, mientras que en la EAIT se centraba en el lóbulo temporal y la unión temporoparietal49,53.
Hasta la fecha, se ha realizado un único estudio longitudinal, según nos consta, comparando la progresión del adelgazamiento cortical en estos grupos, con el hallazgo de una atrofia más rápida y difusa de cortezas asociativas (giros frontales medio e inferior izquierdos, lóbulo parietal inferior izquierdo, parte posterior del giro temporal superior izquierdo, giro fusiforme izquierdo, giros cingulares posteriores bilaterales y precúneo) en la EAIP, y una mayor atrofia comparativa del giro parahipocampal izquierdo en la EAIT54. Este patrón de atrofia se correspondió con el mayor deterioro clínico que presentó el grupo de pacientes con EAIP en funciones ejecutivas, atención y lenguaje54.
Un estudio reciente ha profundizado en la afectación de los núcleos profundos del cerebro en la EAIP y en la EAIT55. En estos pacientes se ha observado una afectación topográfica distinta del estriado. Los pacientes con un inicio precoz presentaron una mayor atrofia del estriado dorsal, mientras que los de inicio tardío tenían una mayor afectación del estriado ventral. Los autores interpretan que estos cambios podrían ser secundarios a la pérdida de aferencias del lóbulo temporal medial, en el caso del estriado ventral, y del parietal en el caso del estriado dorsal. Así mismo, se ha realizado un estudio longitudinal comparando la tasa de atrofia de estructuras subcorticales de los 2 grupos de pacientes56. Se constató una mayor pérdida de volumen del caudado, putamen y tálamo en los pacientes con EAIP comparados con los de EAIT, mientras que la afectación del hipocampo y la amígdala transcurrió paralela entre ambos. Esto se podría relacionar con el hecho de que los pacientes con EAIP muestran un deterioro más rápido en las funciones frontales, pero no en memoria.
Valoración de la sustancia blancaRespecto a la sustancia blanca, se han realizado estudios que han evidenciado una predominante atrofia parahipocampal en la EAIT y un patrón más difuso de atrofia posterior, sobre todo del esplenio del cuerpo calloso y de regiones temporoparietales dorsales en la EAIP, coincidiendo con la topografía de la afectación de sustancia gris, lo que indica que la afectación de la sustancia blanca sería secundaria a la atrofia de la sustancia gris41.
Recientemente, un estudio ha comparado el daño microestructural de la sustancia blanca entre los 2 grupos de pacientes mediante secuencias de difusión tensor (DTI)57. En este se observa una afectación más grave y difusa en el grupo de pacientes con EAIP, en los que se describe una afectación de las conexiones interhemisféricas, sistema límbico y tractos asociativos principales, además del cíngulo posterior, mientras que en la EAIT se afecta, sobre todo, el cuerpo calloso.
En resumen, en la EAIP encontramos una atrofia más difusa tanto de la sustancia gris como de la sustancia blanca, que afecta a áreas neocorticales (sobre todo, el precúneo), mientras que en EAIT se limita al hipocampo. Además, la progresión parece ser más rápida en la EAIP y se extiende a áreas subcorticales, principalmente el putamen.
Neuroimagen funcionalLas técnicas de neuroimagen funcional permiten la valoración de un gran número de funciones cerebrales. Una de las más empleadas es la tomografía por emisión de positrones (PET) con 18F-fluorodeoxiglucosa (FDG), que estima la actividad neuronal mediante la imagen de la tasa de consumo regional de glucosa45. La TC de emisión de fotón único (SPECT) con 99mTc-HMPAO (hexametilpropilenaminooxima marcada con 99mTc), por su parte, estima la actividad neuronal mediante la imagen del flujo sanguíneo cerebral regional45. La RM funcional, por otro lado, nos permite analizar la respuesta cerebral a determinados estímulos, pues se refleja en cambios hemodinámicos en las regiones del cerebro activadas por el estímulo.
Tomografía por emisión de positrones de metabolismo de glucosaEl patrón de afectación típico de la EA en estudios de PET consiste en un hipometabolismo del cíngulo posterior, la corteza temporoparietal posterior y la región anterior de la zona medial de los lóbulos temporales45. El hipometabolismo en dichas regiones resulta más marcado en pacientes con EAIP comparados con los de EAIT, tanto con técnicas de análisis de regiones de interés (analizando estructuras elegidas a priori) como basadas en la vóxeles (en la que se realiza un análisis de la totalidad del cerebro sin necesidad de elegir regiones a priori)45,58,59. Para una misma afectación funcional evaluada por medio del CDR, los pacientes con EAIP presentan un mayor hipometabolismo, lo que se podría atribuir a la mayor reserva funcional de los sujetos jóvenes. Además, la curva de hipometabolismo en relación con el CDR es más pronunciada, indicando una progresión más rápida de la disfunción metabólica59.
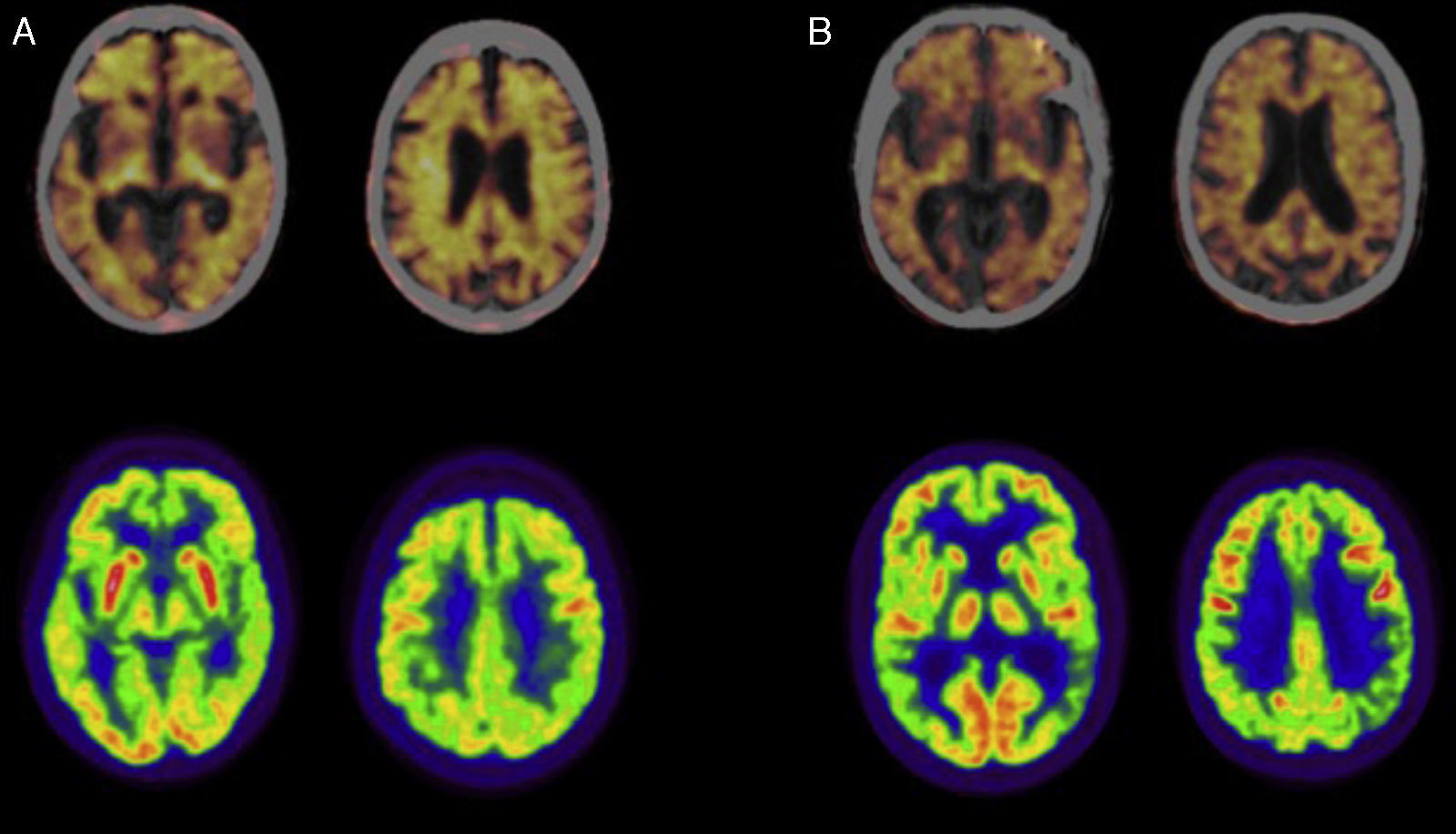
En la figura 1 se muestra el estudio de PET-FDG en un paciente con EAIT y en otro con EAIP (fila inferior).
 con florbetapir (línea superior; imágenes fusionadas con TC) y 18F-Fluorodeoxiglucosa (FDG; línea inferior) en pacientes con diagnóstico de enfermedad de Alzheimer. A) Paciente mujer de 79 años con diagnóstico de enfermedad de Alzheimer típica. La PET con florbetapir es positiva para la presencia de placas de amiloide corticales. El estudio de PET-FDG muestra un hipometabolismo en la corteza de asociación posterior de predominio izquierdo. B) Paciente varón de 57 años con diagnóstico de enfermedad de Alzheimer con afectación predominante del lenguaje. La PET con florbetapir es positiva para la presencia de placas de amiloide corticales. El estudio de PET-FDG muestra un hipometabolismo en la corteza de asociación posterior y de predominio izquierdo.")
Estudios de tomografía de emisión de positrones (PET) con florbetapir (línea superior; imágenes fusionadas con TC) y 18F-Fluorodeoxiglucosa (FDG; línea inferior) en pacientes con diagnóstico de enfermedad de Alzheimer. A) Paciente mujer de 79 años con diagnóstico de enfermedad de Alzheimer típica. La PET con florbetapir es positiva para la presencia de placas de amiloide corticales. El estudio de PET-FDG muestra un hipometabolismo en la corteza de asociación posterior de predominio izquierdo. B) Paciente varón de 57 años con diagnóstico de enfermedad de Alzheimer con afectación predominante del lenguaje. La PET con florbetapir es positiva para la presencia de placas de amiloide corticales. El estudio de PET-FDG muestra un hipometabolismo en la corteza de asociación posterior y de predominio izquierdo.
El patrón esperable en SPECT para la EA típica es similar al descrito para la PET45. Además, los pacientes con EAIP muestran una marcada hipoperfusión de las áreas corticales asociativas posteriores, mientras que los de inicio tardío la tienen en las áreas temporales mediales60.
En resumen, nuevamente encontramos una mayor afectación del neocórtex (parieto-occipital e incluso frontal) en pacientes con EAIP, mientras que los pacientes con EAIT presentan una afectación más restringida al lóbulo temporal medial.
Resonancia magnética funcionalCon esta técnica, cabe destacar un estudio que ha tratado de caracterizar los patrones de conectividad funcional en la EAIP y la EAIT61. El grupo de pacientes con EAIP mostró una disminución de la conectividad de la red prefrontal dorsolateral (DLPFN, relacionada con funciones ejecutivas) y un aumento de la conectividad de la red temporal anterior (ATN, relacionado con la memoria declarativa), mientras que el grupo de pacientes con EAIT presentaba el patrón contrario. Los autores lo relacionan con una topografía de daño neuronal (disminución de conectividad) distinta de un grupo a otro, que desencadena procesos compensatorios (aumento de conectividad).
Tomografía por emisión de positrones amiloideEl desarrollo en los últimos años de radiotrazadores capaces de unirse a la proteína amiloide ha permitido evaluar la presencia de la patología de la EA in vivo45. La mayoría de los estudios realizados con el compuesto Pittsburgh B (PIB) en pacientes con EAIP y EAIT no han mostrado diferencias significativas entre ambos grupos55,62. Para sus autores, la discordancia entre la imagen PET-FDG (como hemos descrito, claramente diferente en ambos grupos) y la de PET-PIB (similar en ambos grupos) indican que tal vez otros procesos patológicos, como la formación de ovillos neurofibrilares, la neuroinflamación o los oligómeros de amiloide (previamente mencionados y no visibles con PET-PIB), sean los responsables de la afectación metabólica, más que el depósito de amiloide fibrilar62. Sin embargo, Ossenkoppele et al. evidenciaron un mayor acúmulo de trazador en el córtex parietal de los pacientes con EAIP63 y un estudio reciente ha mostrado que los paciente con EAIP presentan un mayor acúmulo de amiloide en ganglios basales, tálamo, córtex temporal superior izquierdo y cúneo izquierdo64. Esto sería lo esperable a raíz de la neuroimagen metabólica PET-FDG que ya hemos descrito y teniendo en mente una cascada patológica en que la acumulación de β-amiloide preceda a la disfunción metabólica, aunque quedaría por aclarar qué provoca esa predilección del depósito de amiloide por ciertas áreas63.
De nuevo en la figura 1, también se muestra el estudio de PET de amiloide en un paciente con EAIT y en otro con EAIP (fila superior).
ConclusiónLa EAIP y la EAIT tienen características comunes, pero también muchas diferenciales (tabla 2). El hecho de que el tipo de patología cerebral sea el mismo, siendo este el único criterio de diagnóstico definitivo de la EA, nos obliga a afirmar que técnicamente se trata de una sola entidad. Sin embargo, las grandes diferencias entre una y otra, evidenciadas con este trabajo, nos llevan a cuestionarnos si debe ser únicamente la anatomía patológica la que defina siempre una entidad nosológica cuando se refiere a enfermedades neurodegenerativas como la que nos ocupan.
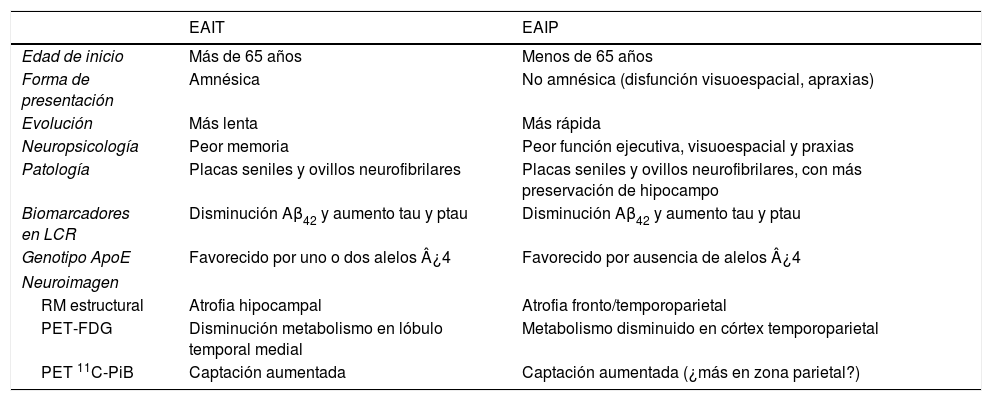
Características generales de la EAIP y la EAIT
| EAIT | EAIP | |
|---|---|---|
| Edad de inicio | Más de 65 años | Menos de 65 años |
| Forma de presentación | Amnésica | No amnésica (disfunción visuoespacial, apraxias) |
| Evolución | Más lenta | Más rápida |
| Neuropsicología | Peor memoria | Peor función ejecutiva, visuoespacial y praxias |
| Patología | Placas seniles y ovillos neurofibrilares | Placas seniles y ovillos neurofibrilares, con más preservación de hipocampo |
| Biomarcadores en LCR | Disminución Aβ42 y aumento tau y ptau | Disminución Aβ42 y aumento tau y ptau |
| Genotipo ApoE | Favorecido por uno o dos alelos ¿4 | Favorecido por ausencia de alelos ¿4 |
| Neuroimagen | ||
| RM estructural | Atrofia hipocampal | Atrofia fronto/temporoparietal |
| PET-FDG | Disminución metabolismo en lóbulo temporal medial | Metabolismo disminuido en córtex temporoparietal |
| PET 11C-PiB | Captación aumentada | Captación aumentada (¿más en zona parietal?) |
Modificado de van del Flier et al.35.
Por otro lado, los resultados hallados en torno de la neuropatología y la neuroimagen lanzan interesantes preguntas acerca de si el proceso fisiopatológico subyacente es o no el mismo en ambos síndromes. Quizás las diferencias entre estas variantes clínicas pudieran explicarse únicamente por los factores relacionados con el envejecimiento. Por otro lado, la aparente mayor agresividad de la EAIP nos obliga a seguir investigando el porqué del diferente comportamiento de esta enfermedad en unos casos y otros.
En resumen, la EAIP muestra con mayor frecuencia una clínica inicial atípica; además, los pacientes evidencian una mayor afectación de la función ejecutiva, visuoespacial y praxias, con menor implicación de la memoria. La EAIP presenta una mayor densidad de patología tipo EA, con una distribución más difusa que la EAIT. Por último, en la EAIP se objetiva un mayor grado de atrofia con afectación más difusa y mayor progresión, que también se observa en las pruebas de imagen funcional con PET y SPECT. Por otro lado, los resultados menos consistentes se encuentran en los estudios de PET que valoran la carga cerebral de amiloide.
Conflictos de interesesNo se declara ninguno.

