



La leucoencefalopatía posthipóxica diferida (LPHD) es una rara complicación que puede ocurrir como consecuencia de un periodo prolongado de hipoxia cerebral.
Se presenta el caso de un varón de 43 años que es encontrado inconsciente en su domicilio. En la exploración se observa una rigidez muscular generalizada, pupilas mióticas y falta de respuesta a estímulos, con una puntuación en la escala de Coma de Glasgow de 3. Inicialmente responde a la administración endovenosa de naloxona, persistiendo importante trabajo respiratorio con taquipnea, taquicardia y roncus difusos en la auscultación. A pesar de recibir oxígeno con FiO2 al 100% y nebulizaciones de salbutamol y bromuro de ipratropio, el paciente desarrolla una insuficiencia respiratoria aguda precisando intubación orotraqueal y ventilación mecánica. Presenta febrícula, y en la placa de tórax hay condensaciones en ambas bases pulmonares, la TAC craneal y la analítica sanguínea inicial son normales. En orina se detecta la presencia de benzodiacepinas, opioides y cannabis. Como antecedentes personales, sufre esquizofrenia paranoide desde la adolescencia, en tratamiento con olanzapina y amisulprida, y es consumidor ocasional de fármacos opioides, sedantes, cannabis y cocaína.
A los 2 días de ingreso, recupera el nivel de consciencia y la exploración neurológica es normal. Como complicación desarrolla una infección respiratoria con buena respuesta al tratamiento antibiótico y un daño renal agudo secundario a la elevación de la enzima creatincinasa (8.277U/l) por probable rabdomiólisis. A los 21 días del ingreso sufre un empeoramiento brusco con somnolencia y bradipsiquia. De forma progresiva va presentando estereotipias y desinhibición motora; el lenguaje es cada vez más escaso con fluencia disminuida y comprensión alterada. Existe una rigidez en rueda dentada en las 4 extremidades. Los reflejos miotáticos son normales y el reflejo cutáneo plantar es flexor en ambos pies. En ningún momento se evidencia déficit focal de fuerza ni sensibilidad. La marcha se deteriora con imposibilidad para caminar, teniendo un gran componente apráxico. Los días siguientes, la clínica empeora hasta quedar en situación de mutismo-acinético.
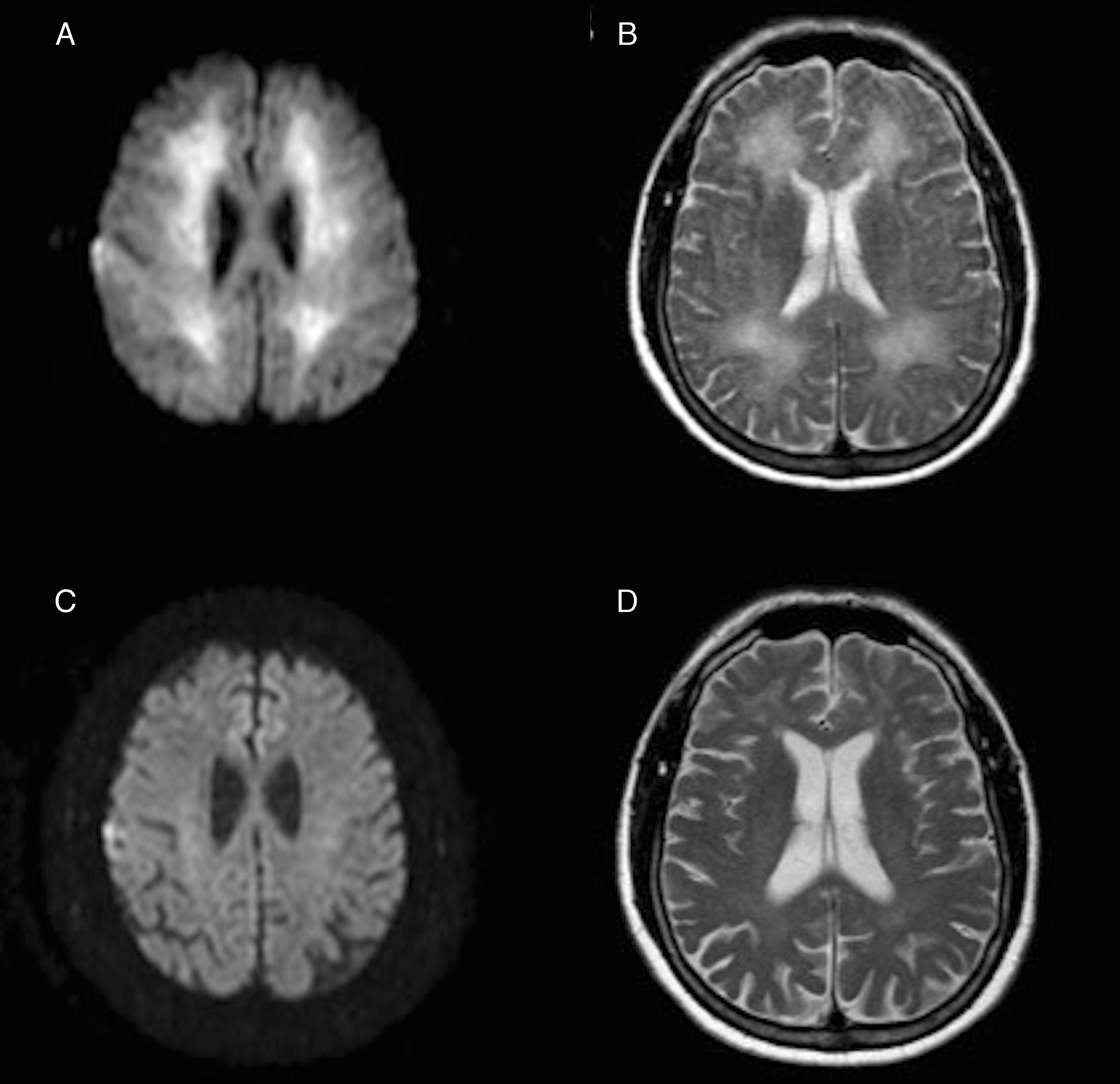
En la resonancia magnética (RM) craneal, realizada 3 semanas después del evento hipóxico, destaca una extensa afectación de sustancia blanca en las secuencias de T2 y FLAIR, y una restricción en las secuencias de difusión (figs. 1A y B).
 e hiperintensidad de señal en la secuencia ponderada en T2 (B) afectando de forma extensa a toda la sustancia blanca periventricular. Debajo, la misma secuencia de difusión (C) y T2 (D), un año después, mostrando la resolución de dichas alteraciones.")
Imágenes axiales de la RM, obtenida 3 semanas después del evento hipóxico, con restricción en la secuencia de difusión (A) e hiperintensidad de señal en la secuencia ponderada en T2 (B) afectando de forma extensa a toda la sustancia blanca periventricular. Debajo, la misma secuencia de difusión (C) y T2 (D), un año después, mostrando la resolución de dichas alteraciones.
El estudio analítico general es normal incluyendo trombofilia, autoinmunidad, serologías, cobre, lactato y niveles de enzima arilsulfatasa A. Es tratado con cuidados generales y rehabilitación, mejora progresivamente, y es dado de alta a los 2 meses y medio en situación basal. Un año después del ingreso, continua asintomático y es independiente para sus actividades diarias. Así mismo, la RM muestra una evolución radiológica muy favorable (fig. 1 C y D). Dada la recuperación clínica y radiológica, y excluidas otras posibles causas inflamatorias o infecciosas, el diagnóstico final es de LPHD reversible.
La LPHD típicamente se caracteriza por un curso bifásico con una recuperación inmediata posterior a un episodio de coma por hipoxia cerebral; seguido el debut de síntomas neuropsiquiátricos días o semanas después de dicho episodio1 a un episodio de coma por hipoxia cerebral; seguido el debut de síntomas neuropsiquiátricos días o semanas después de dicho episodio.
La causa más frecuentemente asociada a esta entidad es la intoxicación por monóxido de carbono2, pero también puede ocurrir tras otros eventos anóxicos como estrangulamientos3, shock hemorrágicos4,5 o sobredosis de opiáceos o sedantes6,7, como en el caso descrito.
Desconocemos el mecanismo fisiopatológico preciso, pero dada la similitud de los hallazgos desmielinizantes en la RM con la leucodistrofia metacromática, un déficit del enzima arilsulfatasa A, que es necesario para el recambio mielínico neuronal, podría predisponer a este síndrome8,9. Se han publicado, sin embargo, varios casos como el nuestro en el que esta enzima presentaba niveles normales10. Otros mecanismos implicados son la toxicidad mieloespecífica de algunos agentes externos11, las alteraciones en la regulación de la vascularización de la sustancia blanca cerebral12 o la susceptibilidad específica de los oligodendrocitos de la sustancia blanca a la hipoxia cerebral13.
El cuadro clínico característico incluye deterioro cognitivo-conductual, desorientación, signos frontales, amnesia, parkinsonismo, mutismo acinético o psicosis. La imagen de RM típicamente muestra una hiperintensidad de señal en secuencias ponderadas en T2 y difusión, y el análisis del líquido cefalorraquídeo es normal. No existe un tratamiento específico, y el pronóstico es variable, pudiendo producirse una recuperación completa, probablemente relacionado con la edad1.
En conclusión, la LPHD es una entidad infrecuente con un cuadro clínico característico típico cuya existencia se debe conocer a fin de evitar tratamientos y pruebas diagnósticas innecesarias, siendo la RM craneal útil para el diagnóstico y el seguimiento evolutivo.
FinanciaciónPara la realización del presente artículo no se ha recibido financiación.
Conflicto de interesesLos autores declaran la ausencia de conflicto de intereses.

