



The incidence of myotonic dystrophy type 1 (DM1), a disease with great phenotypic variety, in our region is unknown. This study aims to estimate the incidence of DM1 at our hospital (a reference centre in Aragon, Spain) and to identify the characteristics of our population (genotype-phenotype correlation).
MethodsRetrospective, descriptive study of 459 patients classified according to the number of CTG repeats, as follows: normal (5–35), premutation (36–50), protomutation (51–80), small expansions (81–150), intermediate expansions (151–1000), and large expansions (> 1000). Furthermore, according to clinical phenotype, patients were categorised as unaffected (5–50 CTG repeats), mild form or asymptomatic (51–150), classical form (151–1000), and severe form (> 1000).
ResultsThe incidence of DM1 was 20.61 cases per million person-years (95% CI, 19.59–21.63). An inverse correlation was observed between the number of CTG repeats and the age at genetic diagnosis (ρ = –0.547; 95% CI, –0.610 to –0.375; P < .001). CTG5 was the most frequent polymorphic allele in healthy individuals. Of all patients with DM1, 28.3% presented the mild or asymptomatic form, 59.1% the classical form, and 12.6% the severe form. Inheritance was maternal in 35.1% of cases, paternal in 59.4%, and uncertain in 5.5%. In mild forms, frontal balding in men was the most prevalent phenotypic trait, as well as myotonia and cataracts, while in the classical form, ptosis, facial weakness, voice and pronunciation alterations, myotonia, and fatigue/sleepiness were most frequent.
ConclusionsThe incidence of DM1 in Aragon is significant. Multidisciplinary study of the phenotype of patients with DM1 is key to early diagnosis and personalised management.
Se desconoce la incidencia de la distrofia miotónica tipo 1 (DM1), enfermedad con gran variedad fenotípica, en nuestra región. El objetivo de nuestro trabajo es estimar la incidencia de DM1 en nuestro centro (referencia en Aragón) e identificar las características propias de nuestra población (correlación genotipo-fenotipo).
MétodosEstudio descriptivo retrospectivo de 459 pacientes clasificados según número de repeticiones CTG en: normal (5–35), premutado (36–50), protomutado (51–80), pequeñas expansiones (81–150), intermedias (151–1000) y grandes (>1000). Además, según el fenotipo mostrado, se categorizaron como: no afectos (5–50 CTGs), forma leve o asintomática (51–150 CTGs), clásica (151–1000 CTGs) y severa (>1000 CTGs).
ResultadosLa incidencia de DM1 fue de 20,61 (IC 95%: 19,59–21,63) casos por millón de individuos-año. Se evidenció una correlación inversa entre el número de CTGs y la edad al diagnóstico genético (ρ = −0,547; IC 95%: −0,610 a −0,375; P <,001). El CTG5 fue el alelo polimórfico más frecuente en sanos. Del total de afectos, 28,3% presentaron la forma leve o asintomática, 59,1% la forma clásica y 12,6% la forma severa. El 35,1% presentaron herencia materna, 59,4% paterna y 5,5% herencia incierta. En las formas leves la calvicie frontal en varones fue el rasgo fenotípico más prevalente junto con miotonía y cataratas, mientras que en la clásica predominó la ptosis palpebral, debilidad facial, alteraciones en la voz y pronunciación, miotonía y sensación de cansancio/somnolencia.
ConclusionesLa incidencia de DM1 es relevante en Aragón. La revisión multidisciplinar del fenotipo de pacientes con DM1 es clave para un diagnóstico precoz y medicina personalizada.
Myotonic dystrophy type 1 (DM1) or Steinert disease (OMIM #160900) is an autosomal dominant myopathy characterised by a CTG trinucleotide repeat expansion in a non-coding region of the DMPK (myotonic dystrophy protein kinase) gene located on the long arm of chromosome 19 (19q13.3).1–4
Due to its great phenotypic variability, every patient needs personalised treatment.5,6 A comprehensive guide to the clinical manifestations of DM1 was published in 2019.5 These include:
- 1
Muscle problems: weakness is predominantly distal, although it can also affect the muscles in the neck and face, as well as those involved in mastication, swallowing, and phonation.7
- 2
Central nervous system manifestations: these are highly variable and include cognitive deficits, apathy, fatigue, sleep disorders,8 and, in cases of neonatal-onset DM1, intellectual disability, attention-deficit/hyperactivity disorder, and executive function disorders.9
- 3
Cardiac symptoms: 75% to 80% of patients present some degree of cardiac involvement, mainly in the form of electrocardiographic alterations and arrhythmia.7,10 The clinical spectrum of these symptoms is variable, ranging from mild electrocardiographic alterations to severe arrhythmia with potential to cause sudden death.3,11,12
- 4
Respiratory alterations: these are frequent among patients with DM1 and constitute one of the main causes of premature death. There seems to be a degree of correlation between the size of the CTG repeat expansion and the severity of respiratory symptoms.13–15
- 5
Skin problems: multiple pilomatricomas, alopecia, seborrheic dermatitis, dysplastic nevus.2
- 6
Endocrine alterations: hypergonadotropic hypogonadism. Men may also present low testosterone levels and elevated levels of FSH and LH. Women may present infertility, miscarriage, and premature ovarian failure.16,17
- 7
Lipid profile alterations and disorders of calcium and phosphorus metabolism: high triglyceride level, low HDL cholesterol level, and vitamin D deficiency (in 90% of patients).
- 8
Gastrointestinal alterations: dysphagia and alterations in mastication due to weakness and myotonia of the muscles of mastication. Other alterations include hypotonic oesophagus, delayed gastric emptying, and alternating periods of diarrhoea and constipation.18
- 9
Ophthalmological alterations: most patients present ocular hypotony and cataracts, which are usually diagnosed after the age of 50 years,19 as well as ptosis secondary to weakness of the levator palpebrae superioris muscle.
- 10
Risks of anaesthesia: anaesthesia with opiates and sedatives should be used with caution, as these patients are particularly sensitive to these drugs.20
Some studies have analysed the prevalence of myotonic dystrophy in different countries21 and various Spanish regions22; however, no study has explored the incidence and prevalence of this disease, or the prevalence of the wide range of associated manifestations, in the adult population of Aragon.
The purpose of this study was to evaluate the incidence of DM1 in the population of Aragon attended by the clinical genetics department of Hospital Universitario Miguel Servet (Zaragoza) between January 2007 and December 2019, by sex, symptoms, and age at genetic diagnosis. Our hospital, a tertiary-level care centre, is the reference centre for genetic testing for DM1 in Aragon. We also evaluated the prevalence of certain clinical traits and studied genotype-phenotype correlations. As secondary objectives, we analysed the type of transmission (maternal/paternal), classified patients by number of CTG repeats into different clinical categories, determined the number of diagnostic and predictive tests conducted, and established the most frequent number of CTG repeats within the normal range.
Material and methodsPatientsWe conducted a retrospective, descriptive study of 459 patients who, during the study period, were referred to our hospital’s clinical genetics department for genetic testing for DM1 (CTG trinucleotide repeat expansion in the DMPK gene).
The inclusion criteria for genetic testing were presence of clinical symptoms or electromyographic findings compatible with DM1 and/or family history of DM1. We gathered data on demographic variables (sex, age at referral for genetic testing, place of residence), number of CTG repeats, clinical manifestations, and family history.
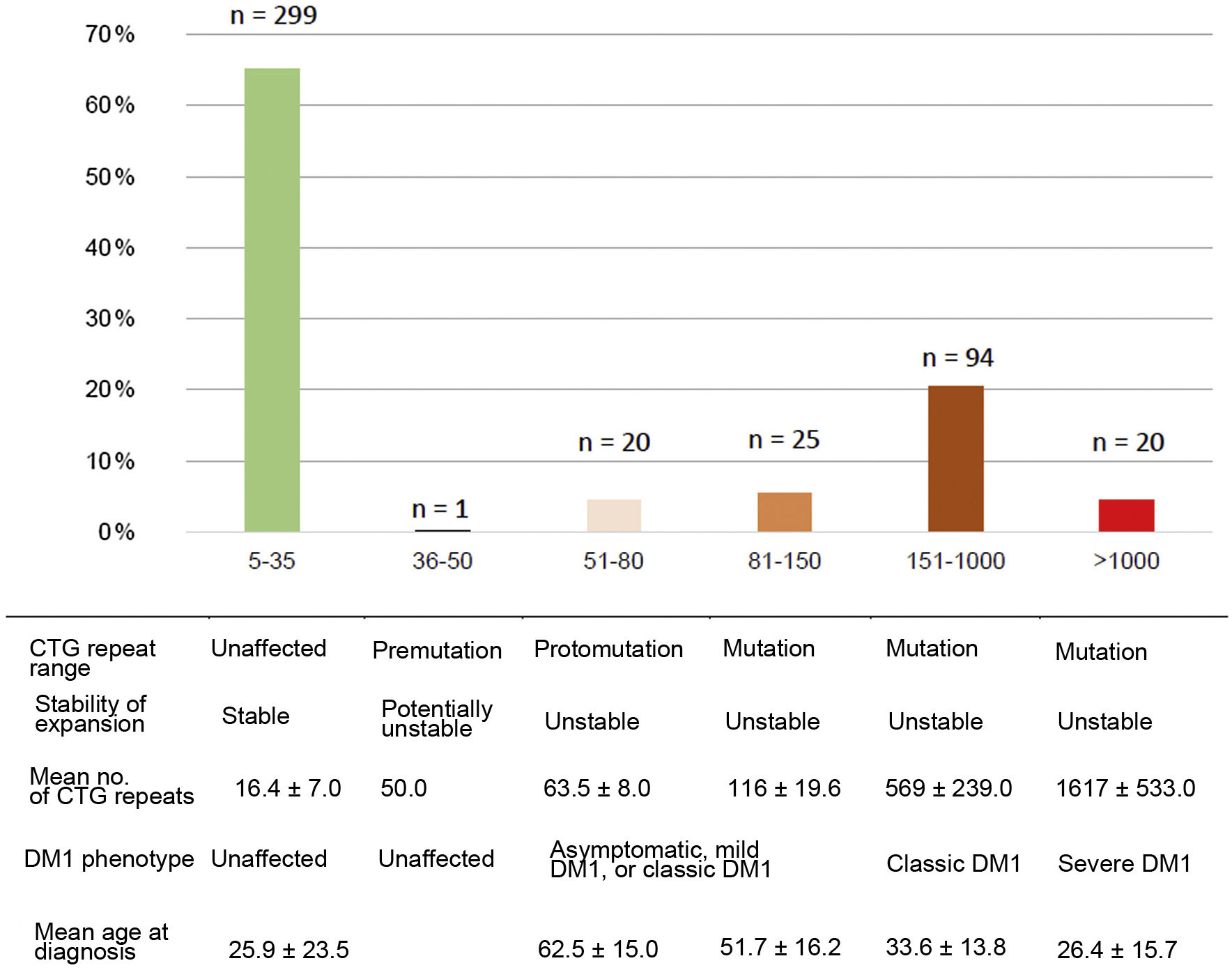
Patients were classified according to the number of CTG repeats into the following categories: normal (5–35 CTG repeats), premutation (36–50), protomutation (51–80), small expansion (81–150), intermediate expansion (151–1000), and large expansion (> 1000 CTG repeats). According to the phenotype, patients were classified into unaffected (5–50 CTG repeats), mild or asymptomatic disease (51–150), classic form (150–1000), and severe disease (> 1000 CTG repeats).2,5,23
SamplesPredictive and symptomatic tests were conducted using peripheral blood DNA in EDTA. For prenatal studies, we used chorionic villus sampling.
Methods and techniquesTwo different techniques were used for genetic testing of DM1 during the study period. Between 2007 and 2011, the number of CTG repeats was determined using conventional PCR and agarose gel electrophoresis, followed by Southern blotting when only one allele could be identified. Conventional PCR has the limitation that it does not provide an accurate estimation of the number of CTG repeats. Between 2012 and 2019, we performed a direct analysis of an unstable CTG nucleotide repeat expansion in the DMPK gene (NM_004409.4) using PCR, capillary electrophoresis, and fluorescent fragment analysis (Adellgene Myotonic Dystrophy Screening kit), and triplet repeat primed PCR (Adellgene Myotonic Dystrophy Confirmatory kit), with an ABI 3130XL sequencer and version 4.0 of the GeneMapper software. These kits have 99% reliability and precision of ± 1 repeat for the ≤ 50 repeat range and ± 3 repeats for the 51–150 repeat range. Our hospital’s clinical genetics department participates in the External Quality Assessment scheme proposed by the European Molecular Genetics Quality Network (EMQN) since 2013, which ensures the accuracy of the results obtained with this technique.
The incidence of DM1 was calculated based on the annual number of newly diagnosed cases per million inhabitants for the period 2007–2019. Estimates of the population attended by our centre each year were provided by the information systems management department.
Statistical analysisWe calculated the frequencies of each clinical category for each qualitative variable. Quantitative variables were analysed with the Shapiro-Wilk test (goodness of fit), and expressed as measures of central tendency (mean or median) and dispersion (standard deviation or percentiles).
Inter-group differences in terms of sex, age at referral, genetic testing results, and family history were analysed with hypothesis contrast testing, comparing proportions (χ2) and means (t test, Mann-Whitney U test), as appropriate. The Spearman correlation coefficient was used to confirm an association between the number of CTG repeats and age at referral. Data were analysed using the Jamovi statistical software, version 1.1.9.0; the threshold for statistical significance was set at P < .05 (two-tailed).
The study was approved by the research ethics committee of the region of Aragon.
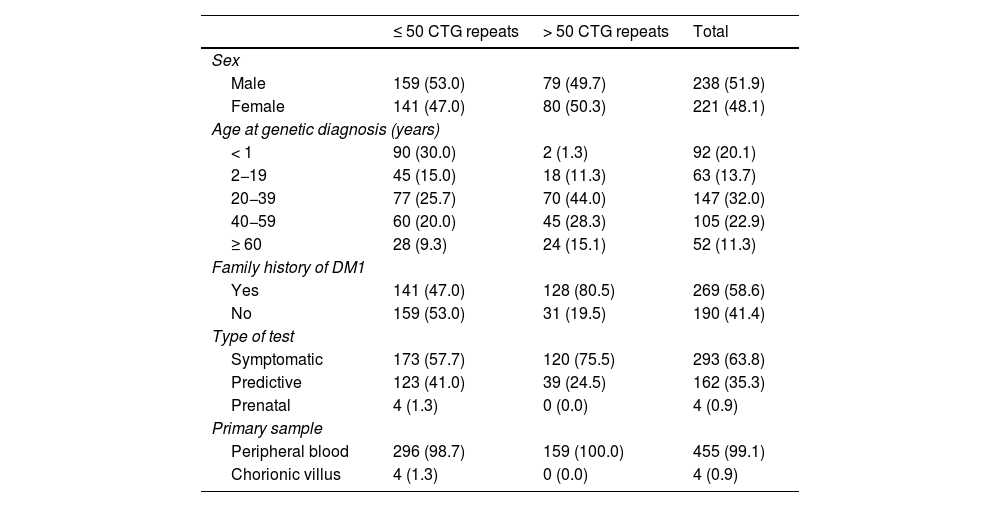
ResultsOf a total of 459 patients included in our study, we identified 159 cases of DM1 (34.6%) (> 50 CTG repeats) in 73 families. The remaining 300 patients (65.4%) were considered unaffected (≤ 50 CTG repeats). Table 1 summarises the demographic characteristics, type of test, clinical form, and family history of patients in both groups.
Demographic characteristics, family history, and results from genetic testing for myotonic dystrophy type 1 in our sample.
| ≤ 50 CTG repeats | > 50 CTG repeats | Total | |
|---|---|---|---|
| Sex | |||
| Male | 159 (53.0) | 79 (49.7) | 238 (51.9) |
| Female | 141 (47.0) | 80 (50.3) | 221 (48.1) |
| Age at genetic diagnosis (years) | |||
| < 1 | 90 (30.0) | 2 (1.3) | 92 (20.1) |
| 2−19 | 45 (15.0) | 18 (11.3) | 63 (13.7) |
| 20−39 | 77 (25.7) | 70 (44.0) | 147 (32.0) |
| 40−59 | 60 (20.0) | 45 (28.3) | 105 (22.9) |
| ≥ 60 | 28 (9.3) | 24 (15.1) | 52 (11.3) |
| Family history of DM1 | |||
| Yes | 141 (47.0) | 128 (80.5) | 269 (58.6) |
| No | 159 (53.0) | 31 (19.5) | 190 (41.4) |
| Type of test | |||
| Symptomatic | 173 (57.7) | 120 (75.5) | 293 (63.8) |
| Predictive | 123 (41.0) | 39 (24.5) | 162 (35.3) |
| Prenatal | 4 (1.3) | 0 (0.0) | 4 (0.9) |
| Primary sample | |||
| Peripheral blood | 296 (98.7) | 159 (100.0) | 455 (99.1) |
| Chorionic villus | 4 (1.3) | 0 (0.0) | 4 (0.9) |
Data are expressed as absolute and relative frequencies (n [%]).
DM1: myotonic dystrophy type 1.
Among patients with > 50 CTG repeats, the male/female ratio was practically 1:1, with 80 affected women (50.3%) and 79 affected men (49.7%). A total of 128/159 affected individuals (80.5%) had family history of clinically suspected or genetically confirmed DM1. Of these, inheritance was maternal in 45 (35.1%), paternal in 76 (59.4%), and uncertain in the remaining 7 (5.5%) (genetic study of the parents was conducted at another laboratory, the parents had died, or recent diagnosis prevented the determination of the inheritance pattern at the time of this study).
Of all genetic studies, 293/459 (63.8%) were performed in patients presenting symptoms compatible with the disease, whereas 162/459 (35.3%) were predictive tests and 4/459 (0.9%) were prenatal tests. Of all the predictive studies performed, 39 (24.5%) yielded positive results, enabling the diagnosis of DM1 in individuals without distinctive symptoms. All 4 prenatal studies yielded negative results.
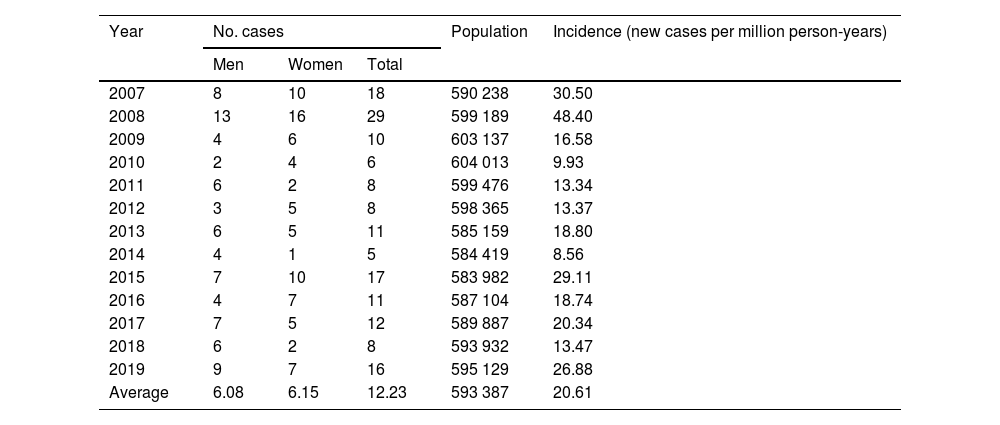
During the 2007–2019 period, the total population served by our hospital’s clinical genetics department was a mean of 593 387 people per year; this translates into an annual incidence of 20.61 cases per million population (calculated based on the number of new cases diagnosed by genetic studies) (Table 2). Similar numbers of positive cases/year were observed in both sexes (6.08 [95% CI, 5.43–6.73] in men vs 6.15 [95% CI, 5.25–7.05] in women; χ2 = 0.410; P = .522).
Number of cases and crude annual incidence of myotonic dystrophy type 1 in Aragon in the period 2007–2019.
| Year | No. cases | Population | Incidence (new cases per million person-years) | ||
|---|---|---|---|---|---|
| Men | Women | Total | |||
| 2007 | 8 | 10 | 18 | 590 238 | 30.50 |
| 2008 | 13 | 16 | 29 | 599 189 | 48.40 |
| 2009 | 4 | 6 | 10 | 603 137 | 16.58 |
| 2010 | 2 | 4 | 6 | 604 013 | 9.93 |
| 2011 | 6 | 2 | 8 | 599 476 | 13.34 |
| 2012 | 3 | 5 | 8 | 598 365 | 13.37 |
| 2013 | 6 | 5 | 11 | 585 159 | 18.80 |
| 2014 | 4 | 1 | 5 | 584 419 | 8.56 |
| 2015 | 7 | 10 | 17 | 583 982 | 29.11 |
| 2016 | 4 | 7 | 11 | 587 104 | 18.74 |
| 2017 | 7 | 5 | 12 | 589 887 | 20.34 |
| 2018 | 6 | 2 | 8 | 593 932 | 13.47 |
| 2019 | 9 | 7 | 16 | 595 129 | 26.88 |
| Average | 6.08 | 6.15 | 12.23 | 593 387 | 20.61 |
Mean (SD) age at genetic diagnosis was 38.9 (18.6) years (range, 0–88), with 44.0% of patients being diagnosed before the age of 35. No statistically significant differences were observed in mean age at genetic diagnosis of DM1 between men (40.7 [20.2]; range, 0–88) and women (37.2 [16.8]; range, 0–77) (t = −1.19; P = .234). Among patients with DM1, mean age at genetic diagnosis was 37.0 (16.1) years (range, 6–63) in those without family history of DM1, and 39.4 (19.2; range, 0–88) in those with family history of the disease (t = 0.638; P = .524).
In our population, the maximum number of CTG repeats was 2800, with a mean of 569.9 (527.7) repeats. No significant differences were found in the number of CTG repeats between individuals with maternal and paternal transmission (U = 1578; P = .520) or between sexes (U = 15 386; P = .216). Lastly, the Spearman correlation coefficient revealed an inverse correlation between the number of CTG repeats and age at genetic diagnosis (ρ = −0.547; 95% CI, −0.610 to −0.375; P < .001).
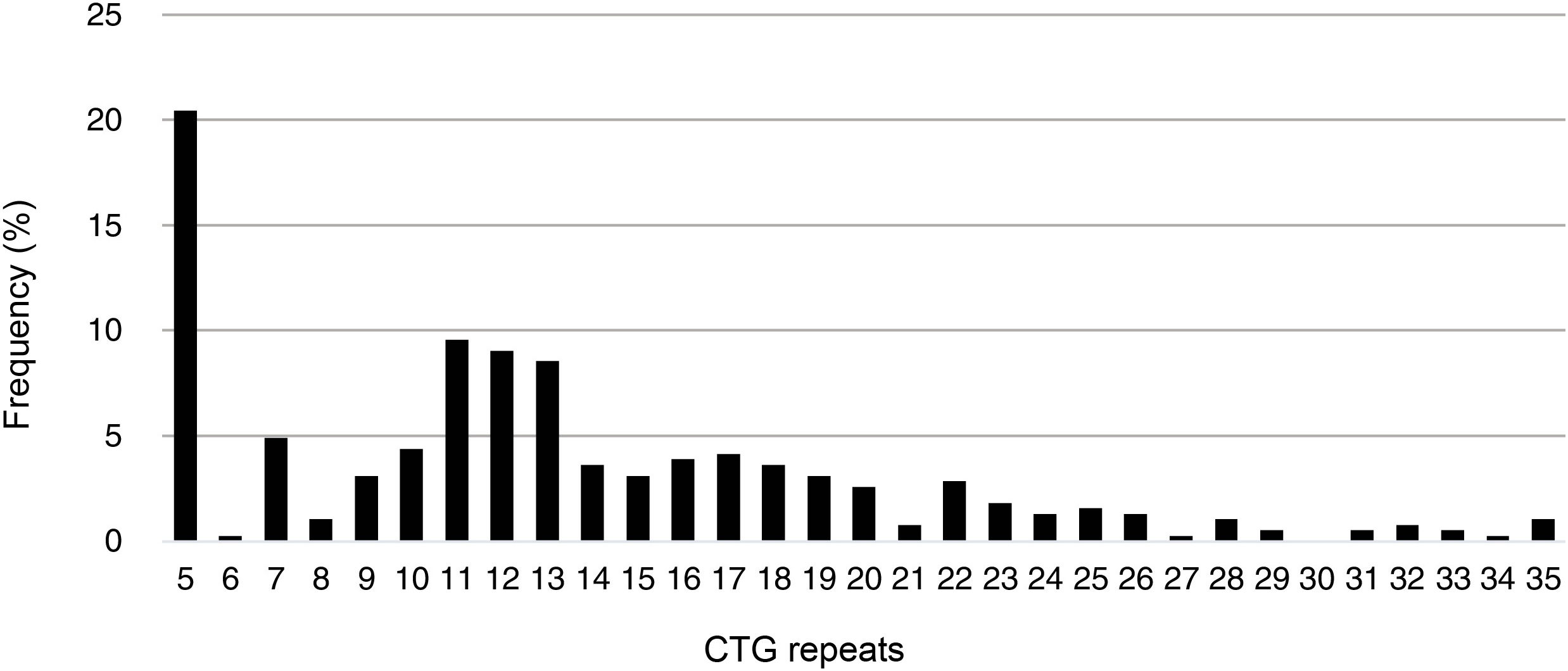
The minimum number of CTG repeats found in the study population was 5 (CTG5); this was also the most frequent number of CTG repeats among unaffected individuals (20.5%), followed by CTG11 (9.6%), CTG12 (9.1%), and CTG13 (8.6%). The mean number of CTG repeats in this group was 13.3 (7.0). Fig. 1 shows the frequencies of different numbers of CTG in unaffected individuals in our population.

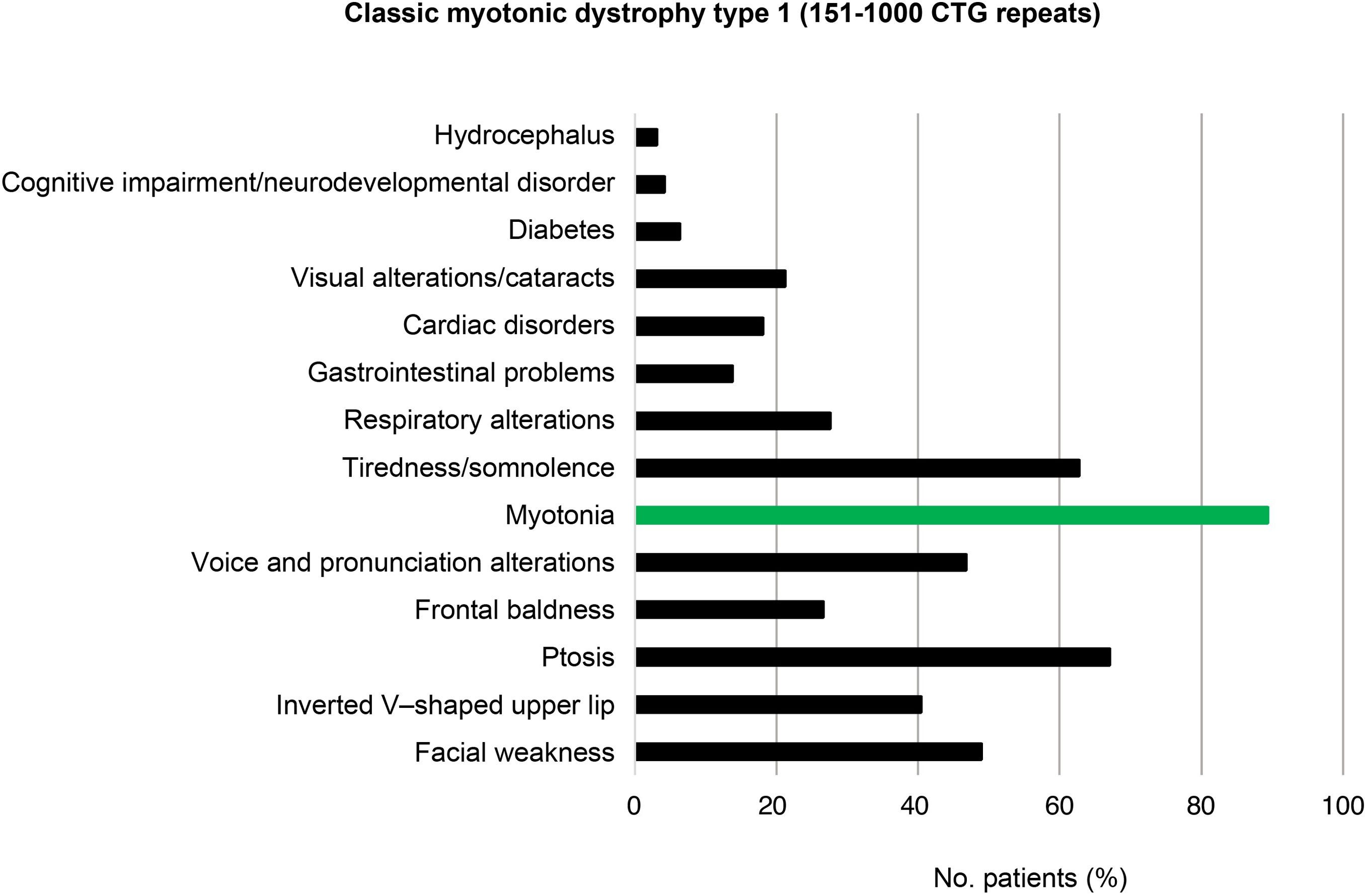
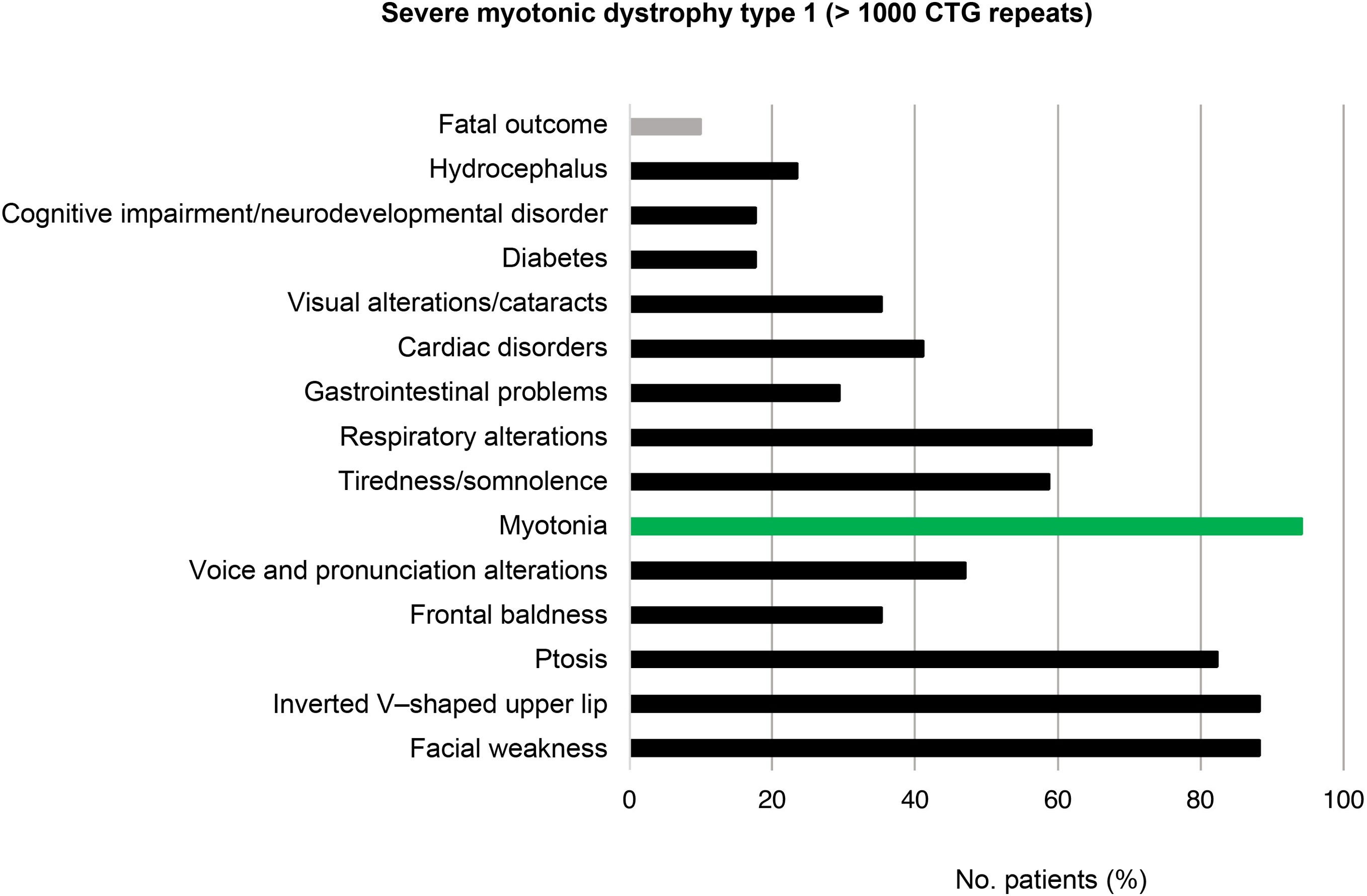
Of the total number of cases of DM1, 45/159 (28.3%) presented mild or asymptomatic disease, 94/159 (59.1%) presented classic DM1, and 20/159 (12.6%) presented severe disease. Fig. 2 presents the clinical and genetic characteristics of our sample, by CTG repeat range.

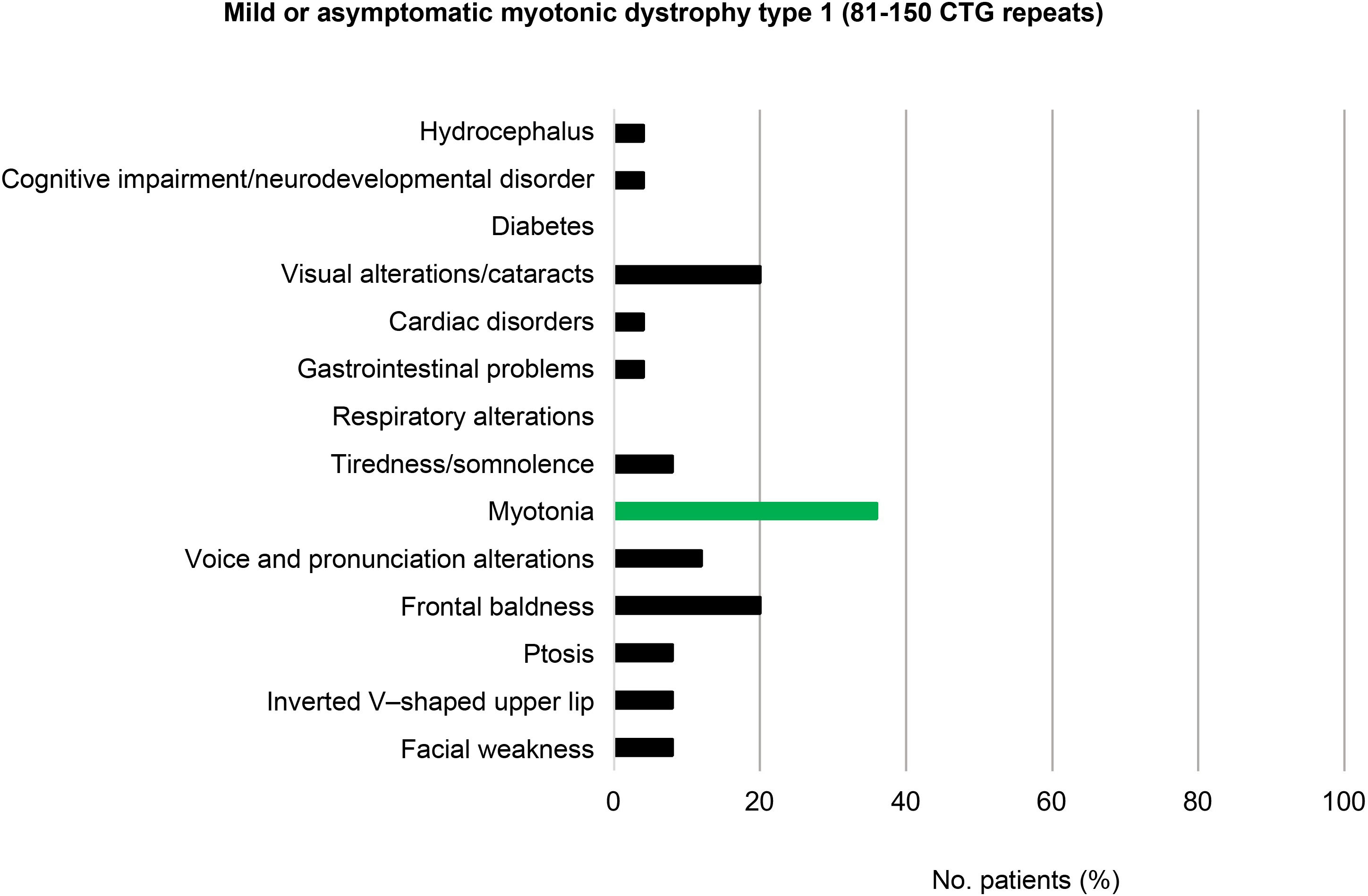
Regarding the type of genetic test performed, 19/45 (42.2%) of patients with mild DM1 were diagnosed with the disease after presenting compatible symptoms, whereas the remaining 26 (57.8%) were diagnosed with predictive tests, but presented no symptoms of DM1 at the time of the study. Of the 94 patients with classic DM1, 84 (89.4%) were diagnosed after undergoing symptomatic testing, whereas the remaining 10 (10.6%) underwent predictive tests.
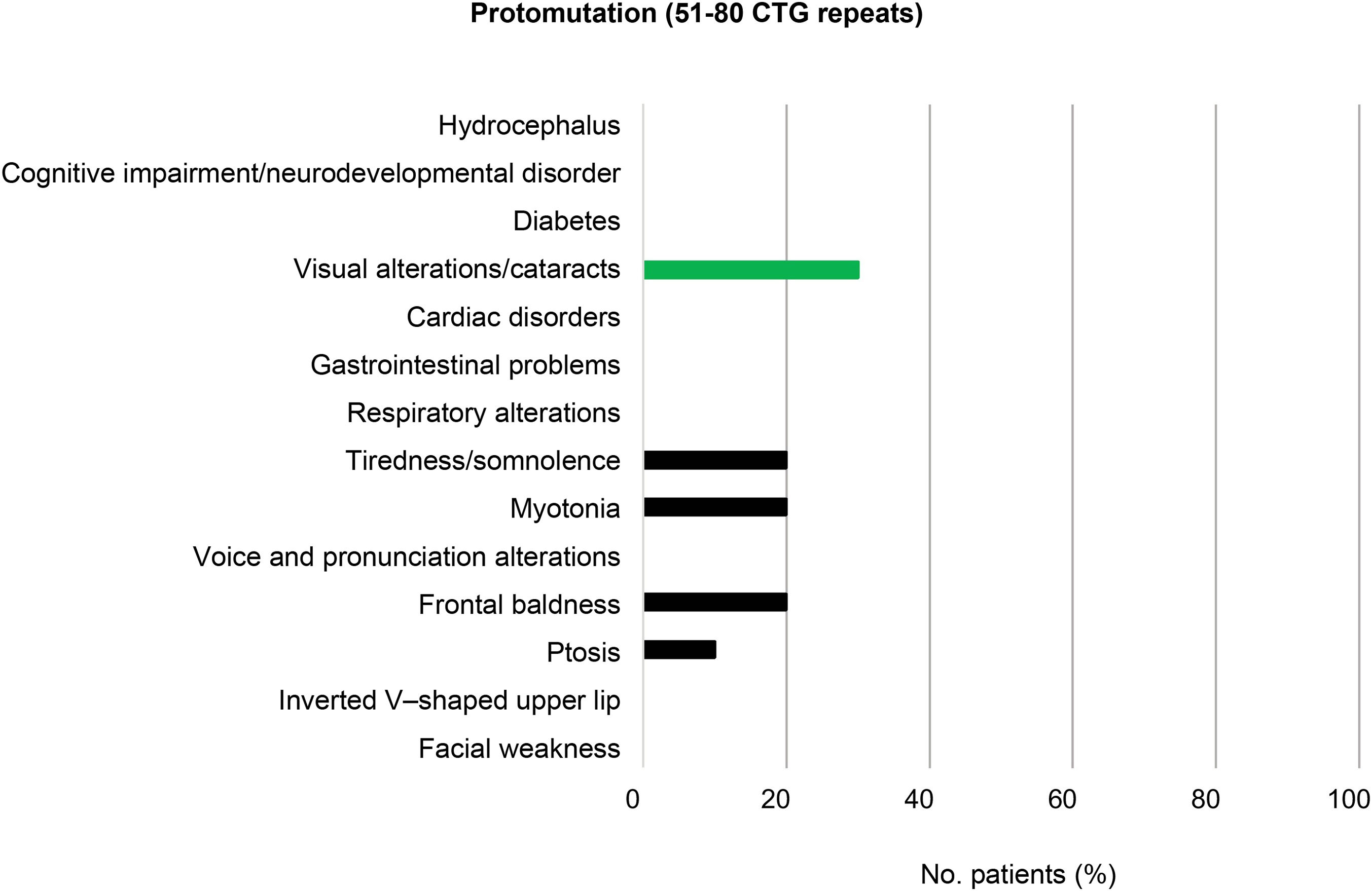
Among the patients with mild or asymptomatic DM1 (81–150 CTG repeats) and those with protomutations (51–80 CTG repeats), the most prevalent phenotypic trait was frontal baldness, present in 20% of individuals in each group. Voice and pronunciation alterations constituted the second most frequent finding among patients with mild DM1 (12%), but were found in none of the protomutation allele carriers.
Visual alterations, myotonia, and tiredness and somnolence complete the typical picture of these patients (20%, 36%, and 8%, respectively, in individuals with mild or asymptomatic DM1, and 30%, 20%, and 20% in protomutation allele carriers). None of the patients in these 2 groups presented respiratory alterations or diabetes (Fig. 3 and 4).
.")
.")
In the 94 patients with classic DM1 (151–1000 CTG repeats), the most frequent features were myotonia (84; 89.4%), ptosis (63; 67.0%), tiredness/somnolence (59; 62.8%), facial weakness (46; 48.9%), and voice and pronunciation alterations (44; 46.8%) (Fig. 5).
.")
Regarding the group of patients with severe DM1 (> 1000 CTG repeats), the clinical characteristics of the disease were reported in 17/20 of the cases. Some of the most prevalent features were facial weakness (15/17; 88.2%), inverted V–shaped upper lip (15/17; 88.2%), and ptosis (14/17; 82.4%). Again, myotonia was the most frequent clinical manifestation (16/17; 94.1%). Presence of motor retardation, respiratory distress, and polyhydramnios were reported in 5 (83.3%) of the 6 patients diagnosed with DM1 before the age of 15 years and with very severe congenital forms of the disease. Two patients with severe DM1 died due to the disease, resulting in a mortality rate of 10.0% (Fig. 6).
.")
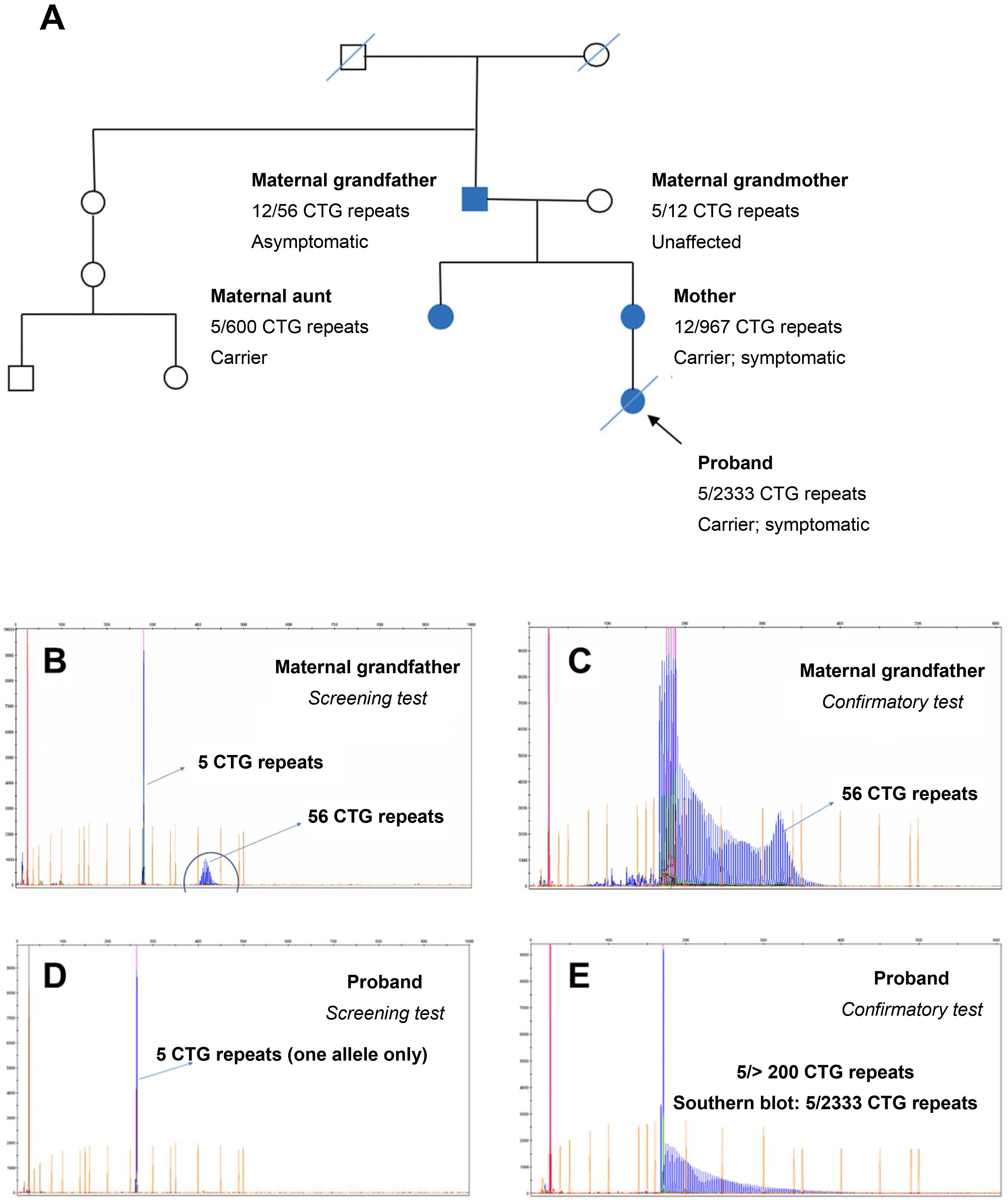
Of all the families participating in the study, we should highlight the one presented in Fig. 7, which presented both paternal and maternal transmission (the proband’s grandfather, a protomutation allele carrier, transmitted the disease to the proband’s mother). According to recent studies,23 protomutation alleles cause greater intergenerational instability; this is observed in the family presented in Fig. 7, in which the 2 daughters of the proband’s grandfather presented 600 and 967 CTG repeats, respectively. The proband presented congenital DM1 due to maternal transmission, and died few months after birth, with hypotonia and severe respiratory distress. Fig. 7 also presents the electropherograms obtained by genetic testing with fluorescence PCR and triplet repeat primed PCR. The electropherograms of the confirmatory tests conducted in the protomutation allele carrier and the proband revealed a similar pattern, confirming the presence of an expanded allele with over 200 CTG repeats; the precise size of the expansion was determined by Southern blot analysis.
. A) Pedigree chart. B,C) Electropherograms for the screening and confirmatory tests performed in the proband’s maternal grandfather. D,E) Electropherograms for the screening and confirmatory tests performed in the proband.")
Study of a family including 2 patients with myotonic dystrophy type 1 (protomutation and full mutation, respectively). A) Pedigree chart. B,C) Electropherograms for the screening and confirmatory tests performed in the proband’s maternal grandfather. D,E) Electropherograms for the screening and confirmatory tests performed in the proband.
In our population, the incidence of DM1 was 20.61 cases per million person-years; this rate is relatively high. Although few studies have calculated the incidence rate of DM1 (with the exception of a study conducted in Belgrade, Serbia,24 which reported a much lower incidence rate, at 2.0 cases per million person-years), several studies do report prevalence rates for DM1 in other Spanish regions (Mallorca: 10.8 cases/100 000 population; northern Spain: 35.9 cases/100 000 population)22 and other predominantly white populations, such as Italy, Israel, northern United Kingdom, Serbia, and western Sweden (9.6–11.7, 15.7, 10.4, 5.3, and 17.8 cases per 100 000 population, respectively).21,25
Among patients presenting a polymorphic allele within the normal range, the most frequent size was 5 CTG repeats, followed by 12 CTG repeats. This has previously been reported in the literature, with alleles with 5 CTG repeats being the most frequent according to several studies.26,27
Among our patients with DM1, the male/female ratio was practically 1:1, suggesting that men and women have the same risk of presenting the disease. This contradicts the findings of some studies that report greater frequency in men.28
Among patients with mild DM1, the most frequent clinical manifestation was myotonia (36%). Mild DM1 is diagnosed at older ages than classic or severe forms of the disease. As a general rule, alleles with larger CTG repeat expansions are associated with greater symptom severity (variable expressivity) and earlier symptom onset (anticipation phenomenon). Mild DM1 is defined as expansions in the range of 81–150 CTG repeats, and is associated not only with mild myotonia but also with frontal baldness (especially in men) and cataracts, with onset at older ages than in patients with more severe forms. In our study, with the exception of a 29-year-old patient with 130 CTG repeats, all affected individuals presented cataracts over the age of 50 years.
The classic form of DM1 was the most common in this study, accounting for 59.1% of all patients with the disease. It presents with a wide range of clinical manifestations, including myotonia, which is more severe than in mild DM1 and results in inability to quickly relax muscles after use (eg, inability to loosen one’s grip after shaking hands). Most patients present the typical facial phenotype, with lack of facial expression (facial weakness), inverted V–shaped upper lip, ptosis,28 low-set ears, and frontal baldness (especially in men). The prevalence of ptosis in our sample (67.0%) is similar to the rates reported in other studies.29 In our sample, 62.8% of patients reported a permanent feeling of tiredness and somnolence/asthenia; this rate is considerably lower than those reported in other studies.30 Respiratory alterations are also frequent (observed in around 28% of our patients with classic DM1), and constitute one of the main causes of death in these patients; some degree of correlation has been observed between CTG repeat length and severity of respiratory involvement in these patients.14 Cardiac involvement is also frequent in patients with large numbers of CTG repeats. The most frequent cardiac manifestations are arrhythmia and conduction disorders (involving either the His-Purkinje system or the atrioventricular node),31 as was also observed in our population. Patients with respiratory and cardiac involvement present alleles with ≥ 500 CTG repeats, which suggests an association between CTG repeat length and risk of respiratory or cardiac alterations.32 Lastly, gastrointestinal problems were observed in a small percentage of our patients. Larger CTG repeat expansions are associated with more aggressive symptoms.32,33 According to the literature, gastrointestinal problems constitute a frequent manifestation of DM1, but have not been well studied and are frequently disregarded by both patients and physicians.2 In our study, 15.4% of patients reported gastrointestinal problems (approximately 5% had mild or asymptomatic forms, 17% had classic DM1, and 28% had severe DM1); however, the percentage of patients with these manifestations is probably much higher.34,35
In our population, the mortality rate for severe and/or congenital forms of the disease was 10.0%; this rate is lower than reported in the literature, with some studies reporting rates as high as 16% to 41%.36,37 The small number of patients with DM1 may explain these discrepancies. In addition to the classical manifestations of the disease, some patients may also present cognitive alterations. In our population, cognitive problems were observed in 19.0% of patients with severe DM1 and 5% of patients with classic DM1. As reported in other studies, hypotonia and respiratory distress are the most prevalent symptoms in congenital forms.38
In our study, all cases of severe DM1 were transmitted maternally. In studies with larger samples, maternal transmission was observed in 90% of all cases of congenital DM1.39 The underlying molecular mechanism by which maternal transmission results in larger numbers of CTG repeats and causes congenital DM1 is unknown. Recent studies suggest that higher levels of methylation cause larger expansions, increasing disease severity.33
Predictive tests, performed in asymptomatic individuals or patients with very mild symptoms who undergo these tests because they have an affected relative, are frequently used in individuals younger than 30 years, as was the case in our population, with the exception of 2 patients (aged 45 and 50 years). In some of these cases, the test detected over 200 CTG repeats, which may be explained by the fact that the disease had not begun to manifest in these patients due to their young age.
Transmission was paternal in 59.4% of cases and maternal in 35.1%. This may be because maternal transmission is associated with larger expansions, leading to more severe or congenital forms of DM1 in offspring, which may cause these patients not to have children. The transmission pattern in the remaining 5.5% of patients is uncertain for several reasons, including recent diagnosis in patients whose parents had not yet undergone genetic testing, patients who had undergone family studies at another laboratory/centre, and patients diagnosed with DM1 whose parents had died before tests could be performed.
The main limitation of the study is the retrospective nature of our series, made up of patients from our hospital’s genetic testing database. As a result, we may have missed older individuals who did not undergo genetic testing, young individuals who did not consent to testing, or individuals with social burdens or without offspring who declined genetic testing. These potential missing data may constitute a source of bias.
In conclusion, the incidence of DM1 in Aragon is noteworthy. Novel diagnostic tools, such as fluorescent PCR and triple repeat primed PCR with Southern blot, should be used to confirm the diagnosis of DM1, obtaining the exact number of CTG repeats and correctly classifying patients according to genotype-phenotype correlations. Larger studies should seek to conduct a thorough clinical evaluation of endocrine and gastrointestinal alterations and other less prevalent disorders in patients with DM1. Multidisciplinary analysis of the clinical characteristics of DM1 is key to early diagnosis and personalised treatment. Genetic testing enables the detection of asymptomatic and mildly symptomatic cases in the families of affected individuals, providing genetic counselling and discussing the available reproductive options.
Conflicts of interestThe authors have no conflicts of interest to declare.
