



Hereditary spastic paraplegias (HSP) are genetic neurodegenerative syndromes presenting a high degree of genetic and clinical heterogeneity, with no clear genotype–phenotype correlation.1–5 The global prevalence of autosomal dominant HSPs ranges from 0.5 to 5 cases/100,000 population, with a mean rate of 1.8 cases/100,000 population. The most prevalent autosomal dominant HSP is spastic paraplegia type 4 (SPG4), caused by mutations in the SPAST gene.6 A recent Spanish epidemiological study reported prevalence rates of 2.24 cases/100,000 population for all HSPs and 0.93 cases/100,000 population for autosomal dominant HSPs, with SPG4 being the most prevalent in the latter group.7 National and international epidemiological studies highlight the need to improve genetic studies, as genetic confirmation is not obtained in up to 50% of cases.
We present the case of a family with HSP in which molecular-genetic analysis identified a new variant of the SPAST gene.
We declare that we followed our centre's protocols on the publication of patient data, and all participants signed the informed consent form.
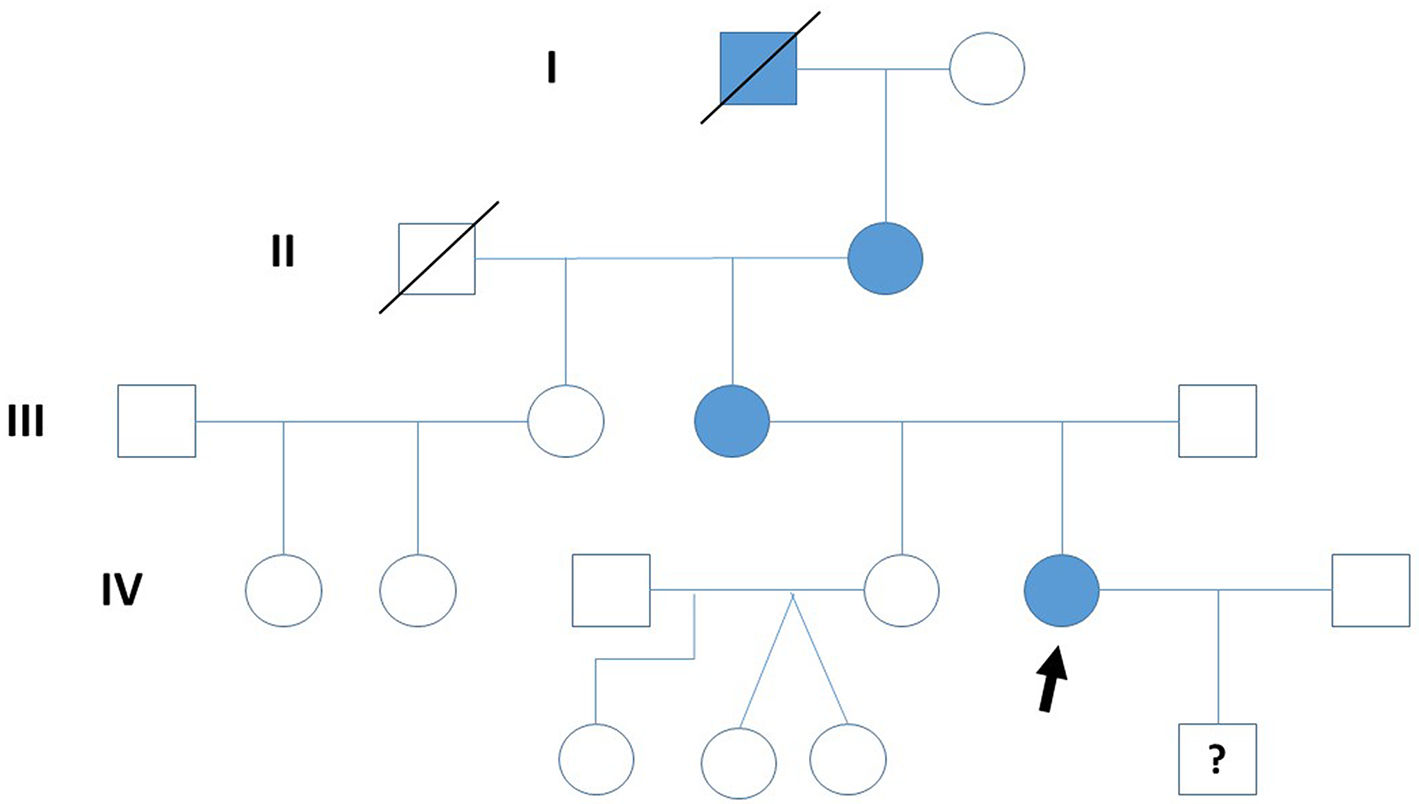
Clinical caseThe study describes four generations of a family, although molecular-genetic analysis was only conducted for the second to the fourth generations. The index case was a patient in the fourth generation of the family (Fig. 1), a 31-year-old woman who has presented a gait disorder since childhood: she tires easily but with effort is able to walk up to 1 km without resting. Neurological examination revealed spastic paraplegia with Achilles tendon clonus and bilateral Babinski, Chaddock, and Hoffmann signs; proprioceptive sensitivity was preserved at the time of examination. We performed complementary testing with a brain and cervical spinal cord MRI study and a neurophysiological study, detecting no relevant alterations. The index patient also presented diabetes mellitus (DM), under treatment with insulin. DM was also present in the index patient’s mother (also treated with insulin) and grandmother (treated with metformin).

Regarding the first generation, we only have documentary evidence of a male patient with a gait disorder and presenting clinical characteristics of spastic paraplegia. In the second generation, the index patient’s grandmother, aged 87 years, presented a gait disorder since adolescence and has used a wheelchair since the age of 70 years; neurological examination revealed spastic paraplegia with bilateral Babinski sign and reduced vibration sensitivity in the lower limbs. In the third generation, the patient’s mother, aged 52 years, presented difficulty walking since childhood. At 12 years of age, she underwent orthopaedic surgery to treat spasticity (adductor and Achilles tenotomy); subsequent disease progression was slow, with the patient using crutches from the age of 45 years. She is currently still able to walk with a walker, although she uses a wheelchair to travel longer distances. Neurological examination revealed bilateral Hoffmann sign, paraplegia with severe spasticity, Achilles clonus, bilateral Babinski and Chaddock signs, and reduced vibration sensitivity in the lower limbs.
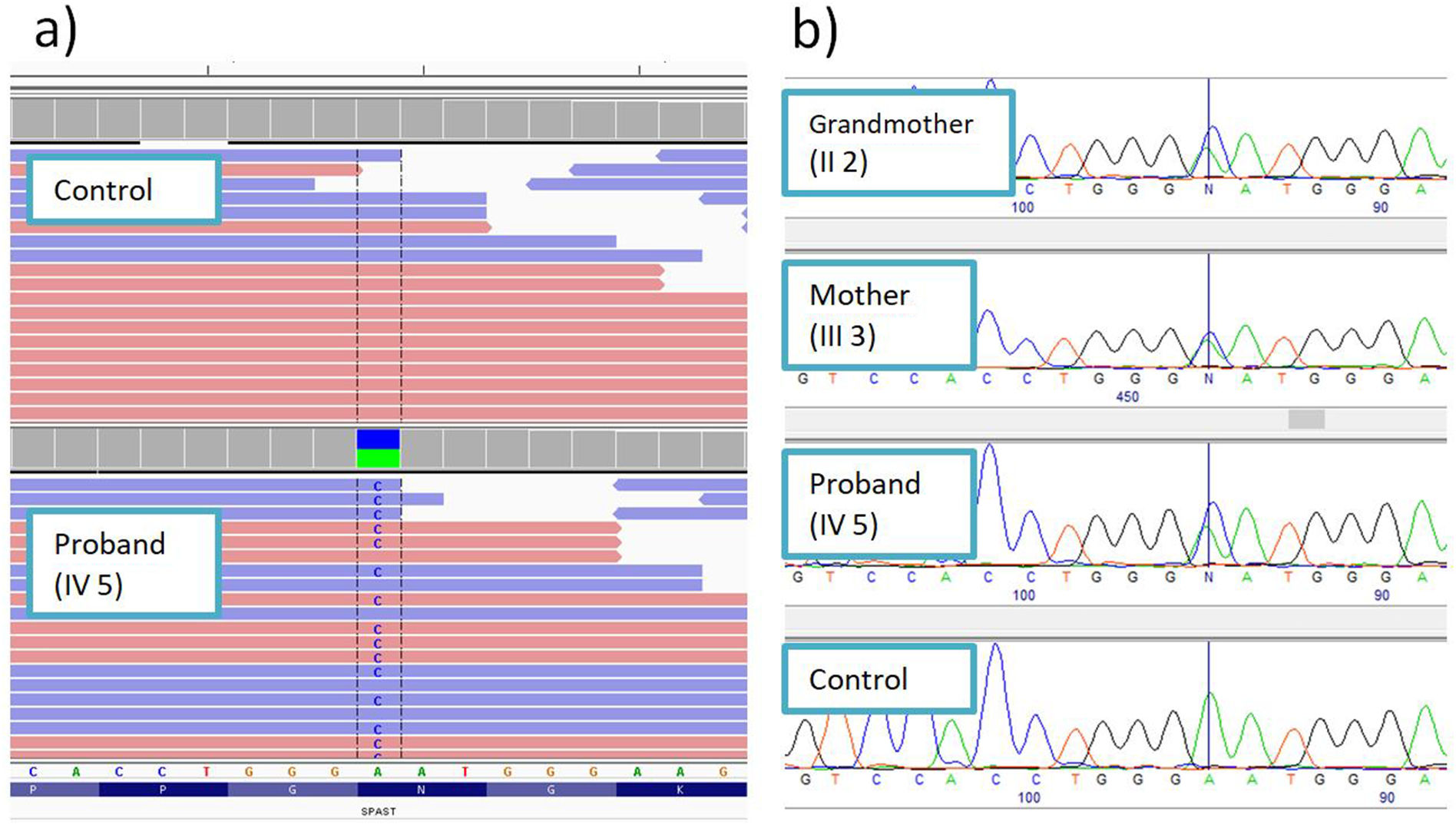
Molecular-genetic studyMassive sequencing of a panel of 155 genes associated with HSP and overlapping syndromes detected a heterozygous variant in exon 8 of the SPAST gene: NM_014946:c.1156A>C (p.Asn386His) (Fig. 2A). This change has not previously been described in the literature and is not reported in population databases. Numerous pathogenic missense variants have been reported in the same amino acid position in patients with HSP: p.Asn386Lys,8 p.Asn386Ser,9 and p.Asn386Tyr.10 The position where the variant was detected is highly conserved at the nucleotide and amino acid levels. All in silico pathogenicity prediction tools classified it as deleterious.
 Variant shown with the Integrative Genomics Viewer. (B) Familial cosegregation analysis conducted by conventional sequencing.")
The familial cosegregation analysis identified the same mutation in the index patient’s mother and grandmother, confirming the autosomal dominant inheritance pattern (Fig. 2B).
DiscussionWe report a family with HSP caused by a novel pathogenic variant of the SPAST gene, c.1156A>C (p.Asn386His). The variant is not correlated with a characteristic clinical phenotype, with the most relevant finding being the presence of diabetes mellitus in all three of the generations studied. Diabetes has very rarely been reported in patients with spastic paraplegia type 7,11 and in two patients from a family with spastic paraplegia type 5,12 and one patient with SPG4.13
We share the goal of the Spanish Society of Neurology’s Commission for the Study of Ataxias and Degenerative Spastic Paraplegias, to increase genetic testing in order to improve the genetic counselling provided to affected families, to raise awareness of these rare diseases, to optimise social and healthcare measures, and to promote clinical trials.7
FundingThis study has not received funding from any source.
Conflicts of interestThe authors have no conflicts of interest to declare.
Supplementary material
