



La enfermedad de Erdheim-Chester (ECD) es una histiocitosis poco frecuente de etiopatogenia desconocida. Su cuadro clínico es muy variado, desde asintomático a marcada afectación del estado general, con sintomatología derivada de la infiltración tisular, habiéndose descrito incluso una forma clínica fulminante con infiltración multisistémica masiva. La principal causa de mortalidad en esta enfermedad es el fallo renal, cardiaco o pulmonar. Presentamos el caso de un paciente que consultó por un cuadro de disnea y tos seca de varios meses de evolución. Inicialmente fue valorado en un centro privado evidenciándose, a nivel radiológico, nódulos pulmonares bilaterales cavitados con moderado derrame pleural bilateral. Tras un estudio inicial que incluyó una toracocentesis, una broncoscopia y una biopsia pleural mediante videotoracoscopia, el paciente fue diagnosticado de probable neumonía organizada criptogenética, iniciándose tratamiento con esteroides a dosis altas, con mejoría sintomática pero con persistencia de las lesiones radiológicas. Por este motivo, el paciente consulta en nuestro hospital para una segunda opinión médica.
Se trata de un varón de 76 años, nunca fumador y con antecedentes de diabetes insípida. En el examen físico presentaba leves crepitantes bibasales y una saturación basal de oxígeno del 94%. En la analítica sanguínea se observó trombopenia leve (105.000 plaquetas/μL) con función renal conservada y patrón de colestasis disociada (GGT 726 U/L, fosfatasa alcalina 205 U/L), siendo el estudio de vasculitis negativo (incluyendo la determinación de anticuerpos anticitoplasma de neutrófilos, antiproteinasa 3, antimieoloperoxidasa y antimembrana basal glomerular). Las pruebas de función respiratoria mostraron una alteración ventilatoria obstructiva moderada con un componente restrictivo asociado. En el ecocardiograma se evidenció una fracción de eyección del ventrículo izquierdo conservada, sin alteraciones en cavidades ni valvulares. En la tomografía por emisión de positrones-tomografía axial computarizada (PET-TAC) se encontraron múltiples opacidades de espacio aéreo en ambos lóbulos superiores, lóbulo medio y língula, con imágenes de broncograma aéreo en su interior. Desde el punto de vista metabólico se apreciaba una captación irregular y multifocal de fluorodesoxiglucosa (FDG) de intensidad leve/moderada (SUVmáx 3,90). Asimismo se observó un pequeño derrame pleural bilateral. Destacaba también la presencia de un aumento de densidad de la grasa mesentérica, con un manguito de tejido de densidad partes blandas, que englobaba a la arteria mesentérica superior y sus ramas, y que se asociaba a un mínimo aumento de la actividad metabólica (SUVmáx 2,52), probablemente inflamatoria. Del mismo modo se evidenció un tejido de similar densidad en el seno de ambos riñones, englobando cálices y pelvis y formando un manguito alrededor del tercio medio uretral (fig. 1). En conjunto, se planteó la posibilidad de una fibrosis. Para completar el estudio se decidió realizar, por parte del Servicio de Cirugía Torácica, una segmentectomía atípica en el lóbulo superior izquierdo y una biopsia de pleura parietal por videotoracoscopia. El estudio anatomopatológico mostró un infiltrado inflamatorio bien delimitado, nodular, que afectaba al intersticio, provocando un ensanchamiento de septos alveolares y de la pleura visceral. El infiltrado inflamatorio era de predominio histiocitario en las zonas más densas y estaba constituido por elementos CD68/Factor XIIIa/CD 163+, siendo negativos los marcadores de mesotelio y epitelio, así como S100, CD 23 y ALK1. La celularidad acompañante correspondía a células linfoides maduras. En la biopsia de pleura parietal se evidenciaron signos de inflamación crónica y fibrosis. Todos estos hallazgos son sugestivos de corresponder a una histiocitosis, concretamente a una ECD (mutación BRAF V600E: negativa).
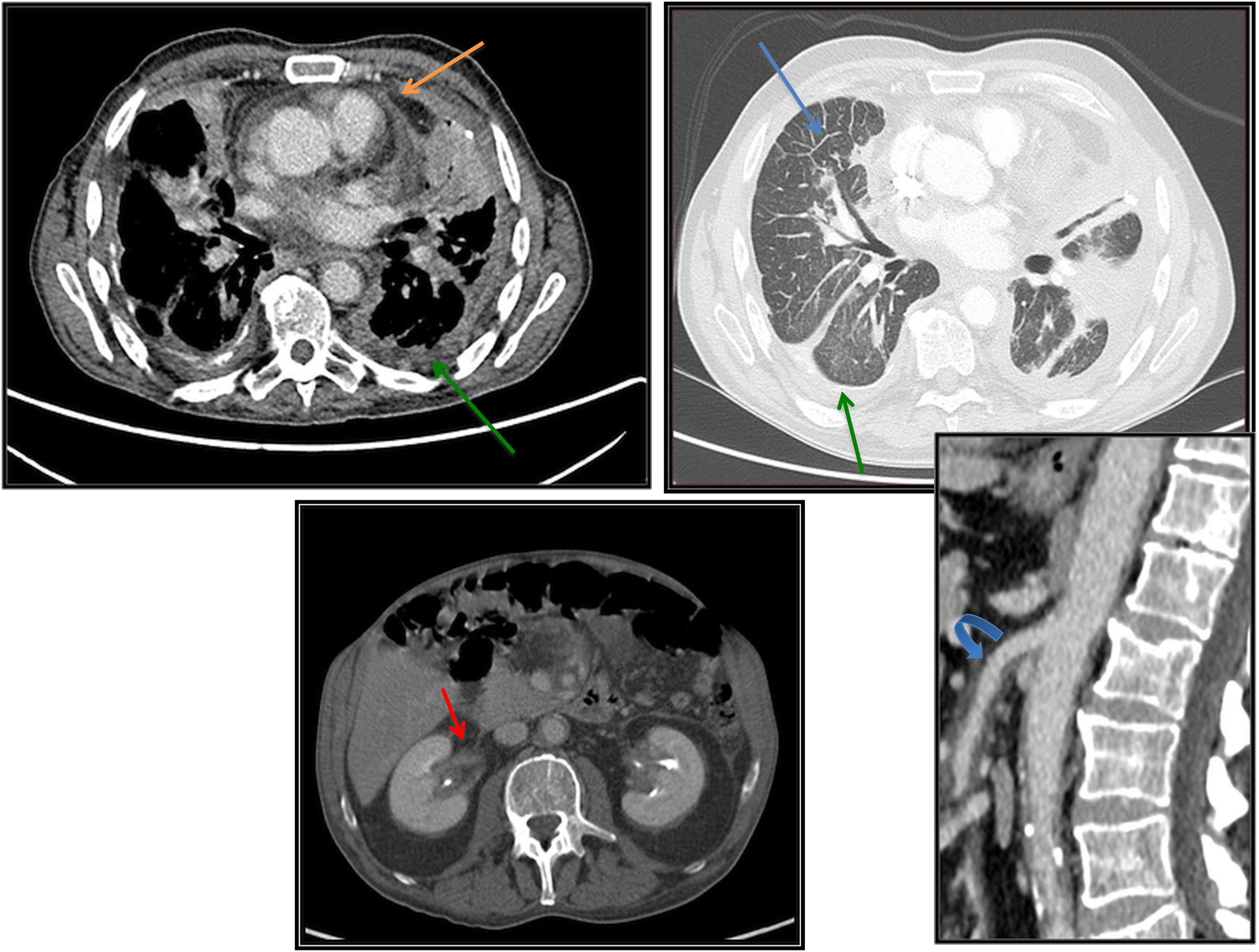
 torácica y abdominopélvica tras la administración de contraste yodado intravenoso. Las imágenes del tórax y la de los riñones son cortes axiales y la de los vasos es una reconstrucción multiplanar en un plano sagital. Se aprecian los siguientes hallazgos: engrosamiento de septos interlobulillares (flecha azul); engrosamiento pleural y de cisuras, bilateral y simétrica, con mínimo derrame pleural (flecha verde); engrosamiento pericárdico con derrame (flecha naranja); engrosamiento mural difuso de las paredes de los vasos de gran y mediano tamaño, como aorta y arteria mesentérica superior (flecha curva azul) que traduce vasculitis; tejido de partes blandas en senos renales y a nivel perirrenal rodeando a la vía urinaria, sin comprimirla (flechas rojas).")
Tomografía axial computarizada (TAC) torácica y abdominopélvica tras la administración de contraste yodado intravenoso. Las imágenes del tórax y la de los riñones son cortes axiales y la de los vasos es una reconstrucción multiplanar en un plano sagital. Se aprecian los siguientes hallazgos: engrosamiento de septos interlobulillares (flecha azul); engrosamiento pleural y de cisuras, bilateral y simétrica, con mínimo derrame pleural (flecha verde); engrosamiento pericárdico con derrame (flecha naranja); engrosamiento mural difuso de las paredes de los vasos de gran y mediano tamaño, como aorta y arteria mesentérica superior (flecha curva azul) que traduce vasculitis; tejido de partes blandas en senos renales y a nivel perirrenal rodeando a la vía urinaria, sin comprimirla (flechas rojas).
La ECD fue descrita por primera vez en 1930 por William Chester y Jakob Erdheim1. Se trata de una enfermedad rara, grupo «L» (histiocitosis de Langerhans)2, caracterizada por la acumulación de macrófagos espumosos, inflamación crónica, fibrosis y disfunción orgánica3. En el año 2016, fue reclasificada por la Organización Mundial de la Salud como una neoplasia hematopoyética de origen histiocítico4.
Aunque puede aparecer en todos los grupos de edad, es más común en los varones de edad adulta. Aunque se especula que esta enfermedad y otros trastornos histiocitarios pueden representar una respuesta aberrante a la infección, hasta el momento no se ha identificado ninguna etiología infecciosa. Tampoco se ha demostrado que se trate de un trastorno genético hereditario5. Su presentación clínica es variable según el sitio afecto. Muchos pacientes tienen manifestaciones óseas al diagnóstico y la gran mayoría presenta también, al menos, un sitio de participación fuera de los huesos. En una serie retrospectiva de 37 pacientes, se evidenció afectación en las siguientes localizaciones6: huesos largos (95%), seno maxilar, vasos grandes y retroperitoneo (59% cada uno), corazón (57%), pulmones (46%), Sistema Nervioso Central (SNC) (41%), piel (27%), glándula pituitaria y órbita (22% cada una). Otros autores han descrito un patrón similar de participación.
La afectación ósea, presente en la mayoría de los pacientes con ECD, suele manifestarse como una osteoesclerosis bilateral y simétrica de la diáfisis de los huesos largos (tibial izquierda en nuestro paciente), afectando también a los del cráneo7, como se detectó en nuestro paciente en una resonancia magnética nuclear (RMN) realizada con posterioridad, en el contexto de su diabetes insípida. La afectación cardiovascular es una fuente significativa de morbimortalidad e incluye anomalías valvulares, engrosamiento pericárdico y fibrosis periaórtica8 (en nuestro paciente estaba afectada la arteria mesentérica superior y sus ramas). La afectación pulmonar, pleural o parenquimatosa, se manifiesta fundamentalmente por tos y/o disnea. A nivel radiológico es característico encontrar un engrosamiento pleural, opacidades nodulares centrilobulillares e incluso infiltración mediastínica9. Las biopsias pulmonares demuestran infiltrados histiocíticos en un patrón linfangítico, con fibrosis asociada, así como infiltrados inflamatorios linfoplasmocíticos10, como en el caso que nos ocupa. En la afectación retroperitoneal es típico encontrar un tejido de densidad de partes blandas que engloba distintas estructuras renales11 (cálices, pelvis y uretra en el caso de nuestro paciente). A nivel del SNC, la afectación hipofisaria se manifiesta comúnmente como diabetes insípida central12, preexistente antes del diagnóstico de ECD en nuestro caso. También puede encontrarse afectación cerebelosa e incluso infiltración de la duramadre, pudiendo llegar a confundirse con un meningioma. Para el diagnóstico de ECD, como ya se ha comentado, es necesaria la biopsia de un tejido afecto. No todos los pacientes requieren tratamiento en el momento del diagnóstico. Éste se reserva para aquellos que presentan enfermedad sintomática, afectación del SNC (predictor independiente de peor pronóstico) o evidencia de disfunción orgánica. En aquellos casos en los que se demuestra la mutación BRAF V600E, se sugiere efectuar un tratamiento inicial con vemurafenib13. En los que no presentan la mutación se recomienda iniciar tratamiento con interferón alfa convencional/pegilado (PEG-IFN)14 o inhibidores MEK (trametinib)15.
Para aquellos pacientes que no toleran o no responden al interferón alfa, se pueden ofrecer otras alternativas de tratamiento con cladribina y ciclofosfamida. El uso de corticoides ha demostrado tener actividad clínica en la ECD, pero no ha conseguido alcanzar beneficios en la supervivencia.
Nuestro paciente recibió tratamiento inicial durante cinco meses con PEG-IFN, que posteriormente se solapó durante cuatro meses más con tocilizumab por empeoramiento pulmonar. En la actualidad el paciente está recibiendo dosis altas de esteroides orales y tocilizumab mensual, habiendo pasado dos años desde el diagnóstico de la enfermedad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
