







Actualmente, la identificación de nuevos casos de déficit de alfa-1 antitripsina (DAAT) continúa siendo uno de los grandes retos a los que se enfrenta la enfermedad. El presente estudio tiene por objetivo realizar un análisis de los resultados de la implementación de un programa de detección de casos sistemático de DAAT para los pacientes con enfermedad pulmonar obstructiva crónica.
Material y métodosEstudio observacional transversal en el que se analizaron los resultados de la detección del DAAT hasta diciembre de 2022. Los casos estudiados se dividieron en tres periodos: 1)sin detección de casos sistemático hasta 2013; 2)detección de casos sistemático de alelos S y Z para casos con AAT <90mg/dl hasta 2018, y 3)detección de casos sistemático de 14 mutaciones para casos con AAT <120mg/dl desde 2018.
ResultadosSe han estudiado un total de 471 casos, de los que 306 (65,0%) eran portadores de alguna mutación relacionada con el DAAT. El número de casos detectados de todas las mutaciones con su porcentaje frente a los estudiados en cada periodo era respectivamente de: 6 (100%), 48 (88,8%) y 253 (61,5%). Si nos limitamos a las mutaciones graves (AAT <57,2mg/dl), la distribución por periodos era respectivamente: 3 (50,0%), 10 (18,5%) y 17 (4,1%).
ConclusionesEl presente estudio describe los cambios en la detección de pacientes portadores de alelos relacionados con el DAAT con tres políticas de identificación de casos diferentes. Los datos avalan la utilización de sistema de detección de casos sistemático en la población de pacientes con EPOC.
Currently, the identification of new cases of alpha-1 antitrypsin deficiency (AATD) continues to be one of the great challenges facing the disease. The present study aims to perform an analysis of the results of the implementation of a systematic case detection program of AATD for patients with chronic obstructive pulmonary disease.
Material and methodsCross-sectional observational study in which the results of AAT screening until December 2022 were analyzed. The cases studied were divided into three periods: (1)no systematic case detection until 2013; (2)systematic case detection of S and Z alleles for cases with AAT <90mg/dL until 2018, and (3)systematic case detection of 14 mutations for cases with AAT <120mg/dL since 2018.
ResultsA total of 471 cases were studied, of which 306 (65.0%) were carriers of some mutation related to HAD. The number of detected cases of all mutations with their percentage against those studied in each period was respectively: 6 (100%), 48 (88.8%) and 253 (61.5%). If we limit to severe mutations (AAT <57.2mg/dL), the distribution by periods was respectively: 3 (50.0), 10 (18.5%) and 17 (4.1%).
ConclusionsThe present study describes the changes in the detection of patients carrying DAAT-related alleles with three different case identification policies. The data support the use of systematic case detection system in the COPD patient population.

A pesar de que los documentos de recomendaciones sobre el diagnóstico y el tratamiento de la enfermedad pulmonar obstructiva crónica (EPOC) indican claramente la necesidad de hacer una detección de casos sistemático del déficit de alfa-1 antitripsina (DAAT) a todos los casos1, actualmente la identificación de nuevos casos de DAAT continúa siendo uno de los grandes retos a los que se enfrenta la enfermedad2.
La importancia de detectar nuevos casos de DAAT es clave por varios motivos: 1)el DAAT no diagnosticado tiene un claro impacto en los pacientes3; 2)el tratamiento aumentativo puede retrasar la pérdida de densidad pulmonar pero no puede recuperar lo ya perdido4, por lo que es necesario comenzar el tratamiento cuanto antes, y 3)es necesario establecer el diagnóstico para poder llevar a cabo una detección de casos familiar adecuado que busque la identificación de casos que permitan una intervención precoz5. Sin embargo, a pesar de la disponibilidad de resultados de cribados en distintos países6-9, en España la implementación de un programa de detección de casos sistemático a los pacientes con EPOC parece que aún está lejos de la práctica clínica en la vida real10.
En este contexto, resulta necesario comunicar los resultados de programas de detección de casos en España de manera que sirva de ejemplo y de estímulo para que otras unidades realicen una implementación de un sistema de detección de casos para los pacientes con EPOC. En nuestro centro, hemos venido realizando un programa de detección de casos sistemático de los pacientes con EPOC desde el año 2014. Durante este tiempo hemos adquirido experiencia sobre el diagnóstico del DAAT y la implementación de un programa de identificación sistemática de estos casos. El presente estudio realiza un análisis de los resultados de la implementación de nuestro programa de detección de casos sistemático para los pacientes con EPOC. Nuestros resultados apoyan sin fisuras la realización de este programa sistemático y ayudarán al clínico a tener datos que sustenten este programa en todos los centros de España.
Material y métodosEstudio observacional transversal con recogida prospectiva de la información en el que se analizaron los resultados de un protocolo de detección de casos para el DAAT en toda la historia del Servicio de Neumología hasta diciembre de 2022. Los pacientes provenían de una consulta monográfica de EPOC de un hospital regional universitario. Los casos estudiados se dividieron en tres periodos según el grado de implementación del protocolo actual. Un primer periodo en el que no se hacía detección de casos sistemático como grupo comparador hasta el año 2013. Un segundo periodo, desde 2014 a junio de 2018, en el que se hacía detección de casos sistemático con determinación de la concentración sérica de alfa-1 antitripsina (AAT) a todos los pacientes con EPOC con estudio de las mutaciones S y Z a los casos con una concentración sérica de AAT <90mg/dl mediante el circuito de REDAAT-SEPAR por estudio de gota seca11. En el tercer periodo, desde mediados de 2018, se implementa el sistema actual REDAAT-Progenika12 con detección sistemática de 14 mutaciones en pacientes con AAT <120mg/dl.
Este protocolo actual de detección de casos implementado en el tercer periodo se resume en la figura 1. Brevemente, en el presente protocolo de detección de casos consiste en la determinación en sangre de la concentración de AAT, junto con la de proteínaC reactiva (PCR). En el caso de presentar una PCR elevada por encima de 5mg/l, se le realizada una segunda determinación de AAT en una visita subsiguiente. Dentro del protocolo se les proponía a los pacientes hacerles un estudio genético para ver genotipos del DAAT a aquellos sujetos que cumplan uno de estos criterios: 1)tener una concentración <120mg/dl de AAT en sangre; 2)tener una elevación de la PCR en sangre en al menos dos determinaciones por encima de 5mg/l; 3)ser familiar de primer grado de un caso ya diagnosticado de DAAT, y 4)presentar una clínica (respiratoria o hepática) muy sugestiva de DAAT a juicio del clínico responsable y no disponer de la concentración sérica de AAT.

Los datos clínicos se recogían de manera prospectiva mediante un cuestionario estandarizado que recogía información sobre: datos sociodemográficos (edad, sexo), factores de riesgo (exposición a tóxicos inhalados), comorbilidades (mediante el cálculo del índice de comorbilidad de Charlson)13, datos de impacto clínico, tratamientos inhalados y orales administrados para su proceso respiratorio, resultado de las pruebas de función respiratoria y datos analíticos (AAT, PCR, hemograma, datos de afectación hepática). Se consideró que había una deficiencia grave de AAT cuando los valores hemáticos de AAT estaban por debajo del considerado actualmente como umbral de protección de 11μM, que corresponden a 57,2mg/dl14.
Los datos del estudio se recopilaron y gestionaron mediante las herramientas de captura electrónica de datos Research Electronic Data Capture (REDCap) alojadas en la Sociedad Española de Neumología y Cirugía Torácica (SEPAR)15,16. REDCap es una plataforma de software segura, basada en la web, diseñada para apoyar la captura de datos para estudios de investigación, y que proporciona: 1)una interfaz intuitiva para la captura de datos validados; 2)registros de auditoría para el seguimiento de la manipulación de datos y procedimientos de exportación; 3)procedimientos de exportación automatizados para descargas de datos sin problemas a paquetes estadísticos comunes, y 4)procedimientos para la integración de datos y la interoperabilidad con fuentes externas.
El estudio fue aprobado por el comité de ética de nuestro centro y todos los pacientes firmaban un consentimiento informado por escrito antes de someterse al estudio genético de DAAT. El análisis de los datos se realizó mediante el paquete estadístico IBM SPSS Statistics (IBM Corporation, Armonk NY, EE.UU.), versión 28.0. Los datos descriptivos se caracterizaron mediante las frecuencias absolutas y relativas de las variables categóricas y mediante la media y la desviación estándar entre paréntesis para las variables continuas.
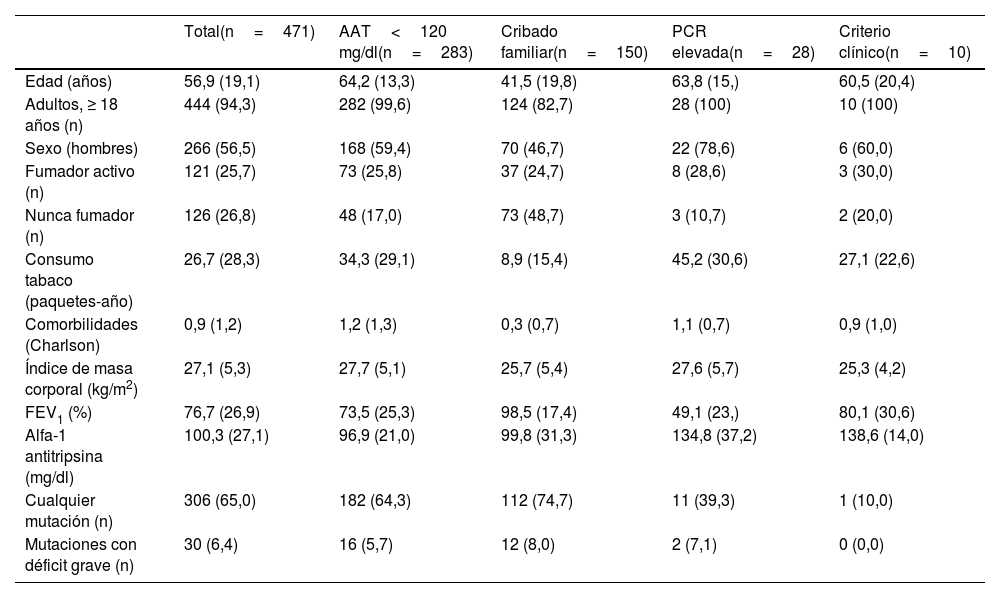
ResultadosHasta diciembre 2022 se han estudiado por sospecha de DAAT un total de 471 casos, de los que 306 (65,0%) eran portadores de alguna mutación relacionada con el DAAT. Los motivos para hacer el estudio genético de DAAT fueron AAT <120mg/dl en 283 (60,1%) casos, cribado familiar en 150 (31,8%) casos, elevación de la PCR en 28 (5,9%) casos, clínica sugestiva a decisión del clínico en 10 (2,1%) casos. Las características basales de los pacientes en global y según el motivo de estudio se resumen en la tabla 1. El rango de edad iba desde 1 hasta 91años. La muestra incluía 26 (5,5%) menores de edad (<18años) incluidos por cribado familiar.
Descripción de la muestra según el criterio para hacer el estudio de genotipado
| Total(n=471) | AAT<120 mg/dl(n=283) | Cribado familiar(n=150) | PCR elevada(n=28) | Criterio clínico(n=10) | |
|---|---|---|---|---|---|
| Edad (años) | 56,9 (19,1) | 64,2 (13,3) | 41,5 (19,8) | 63,8 (15,) | 60,5 (20,4) |
| Adultos, ≥ 18 años (n) | 444 (94,3) | 282 (99,6) | 124 (82,7) | 28 (100) | 10 (100) |
| Sexo (hombres) | 266 (56,5) | 168 (59,4) | 70 (46,7) | 22 (78,6) | 6 (60,0) |
| Fumador activo (n) | 121 (25,7) | 73 (25,8) | 37 (24,7) | 8 (28,6) | 3 (30,0) |
| Nunca fumador (n) | 126 (26,8) | 48 (17,0) | 73 (48,7) | 3 (10,7) | 2 (20,0) |
| Consumo tabaco (paquetes-año) | 26,7 (28,3) | 34,3 (29,1) | 8,9 (15,4) | 45,2 (30,6) | 27,1 (22,6) |
| Comorbilidades (Charlson) | 0,9 (1,2) | 1,2 (1,3) | 0,3 (0,7) | 1,1 (0,7) | 0,9 (1,0) |
| Índice de masa corporal (kg/m2) | 27,1 (5,3) | 27,7 (5,1) | 25,7 (5,4) | 27,6 (5,7) | 25,3 (4,2) |
| FEV1 (%) | 76,7 (26,9) | 73,5 (25,3) | 98,5 (17,4) | 49,1 (23,) | 80,1 (30,6) |
| Alfa-1 antitripsina (mg/dl) | 100,3 (27,1) | 96,9 (21,0) | 99,8 (31,3) | 134,8 (37,2) | 138,6 (14,0) |
| Cualquier mutación (n) | 306 (65,0) | 182 (64,3) | 112 (74,7) | 11 (39,3) | 1 (10,0) |
| Mutaciones con déficit grave (n) | 30 (6,4) | 16 (5,7) | 12 (8,0) | 2 (7,1) | 0 (0,0) |
Datos expresados con media (desviación estándar) o frecuencias absolutas (relativas) según la naturaleza de las variables.
AAT: alfa-1 antitripsina; FEV1: volumen espirado forzado en un segundo; PCR: proteína C reactiva.
La distribución de los diagnósticos en el tiempo está reflejada en la figura 2. El número de casos detectados de todas las mutaciones con su porcentaje frente a los estudiados en cada periodo era respectivamente de: 6 (100%), 48 (88,8%) y 253 (61,5%). Si nos limitamos a las mutaciones graves (AAT <57,2mg/dl), la distribución por periodos era respectivamente: 3 (50,0%), 10 (18,5%) y 17 (4,1%).
 no grave. En azul, el número de casos con DAAT grave.")
La frecuencia de las mutaciones encontradas y su relación con la concentración sérica de AAT está reflejada en la figura 3. En 23 (4,8%) casos no se realizó determinación sérica de AAT en el proceso de estudio, principalmente en niños pequeños evaluados por cribado familiar. La combinación de alelosM con raros eran 2PI*MI, 6PI*MMmalton, 1PI*MPlowell y 1PI*M con una nueva mutación no descrita. La combinación del aleloS con raros eran: 2PI*SI, 1PI*SMheerlen y 2PI*SMmalton. La combinación de alelos Z con raros eran: 1PI*ZI, 3PI*ZMmalton y 1PI*ZMsevilla. La sistematización del estudio del DAAT permitió identificar dos mutaciones nuevas, una con capacidad patogenética, bautizada como PI*Msevilla17, y otra, localizada en un intrón, sin clara implicación patogenética. Si hubiéramos utilizado el punto de corte de 90mg/dl en lugar de 120mg/dl, el número de casos detectados hubiera sido considerablemente inferior, aunque no hubiera afectado al número de mutaciones graves. En la tabla 1 se muestra, además, el número de mutaciones encontradas según el motivo de estudio. Los motivos con más detección de casos eran el estudio sistemático con AAT <120mg/dl y el cribado familiar. El criterio clínico exclusivamente no identificó ninguna mutación grave.
 (90-120mg/dl), el punto de corte para el estudio genético (120mg/dl) y el punto de corte para una deficiencia grave (57,2mg/dl).")
Concentración sérica de alfa-1 antitripsina, según genotipos.
Las líneas discontinuas horizontales representan los valores de normalidad de alfa-1 antitripsina (AAT) (90-120mg/dl), el punto de corte para el estudio genético (120mg/dl) y el punto de corte para una deficiencia grave (57,2mg/dl).
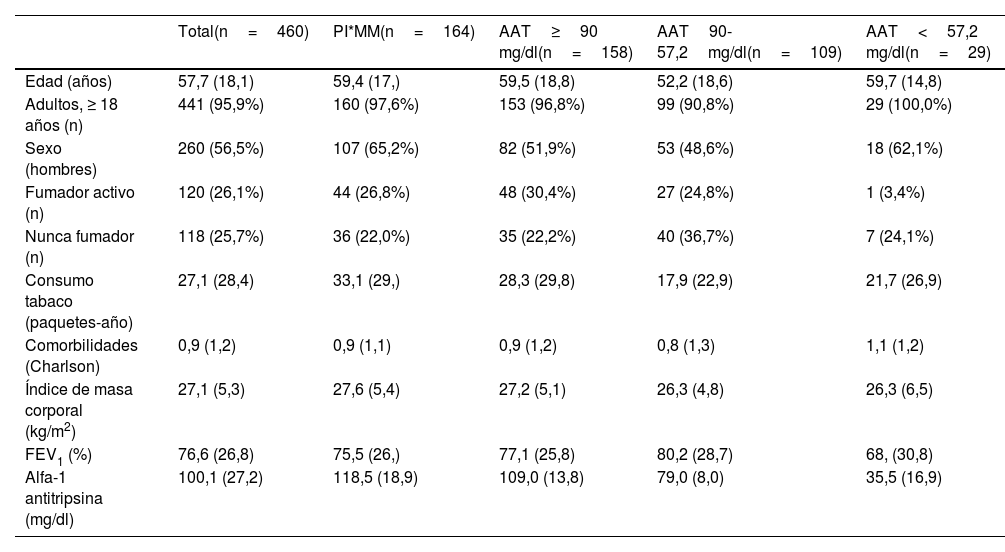
Finalmente, las características clínicas según los grupos de mutaciones y según la gravedad de las mismas están reflejadas en la tabla 2. Aunque algunos grupos presentaban algunas diferencias esperables, realmente ninguna de las características exploradas permitía la identificación de algunos de los grupos.
Descripción de la muestra según las mutaciones y la gravedad del DAAT
| Total(n=460) | PI*MM(n=164) | AAT≥90 mg/dl(n=158) | AAT90-57,2mg/dl(n=109) | AAT<57,2 mg/dl(n=29) | |
|---|---|---|---|---|---|
| Edad (años) | 57,7 (18,1) | 59,4 (17,) | 59,5 (18,8) | 52,2 (18,6) | 59,7 (14,8) |
| Adultos, ≥ 18 años (n) | 441 (95,9%) | 160 (97,6%) | 153 (96,8%) | 99 (90,8%) | 29 (100,0%) |
| Sexo (hombres) | 260 (56,5%) | 107 (65,2%) | 82 (51,9%) | 53 (48,6%) | 18 (62,1%) |
| Fumador activo (n) | 120 (26,1%) | 44 (26,8%) | 48 (30,4%) | 27 (24,8%) | 1 (3,4%) |
| Nunca fumador (n) | 118 (25,7%) | 36 (22,0%) | 35 (22,2%) | 40 (36,7%) | 7 (24,1%) |
| Consumo tabaco (paquetes-año) | 27,1 (28,4) | 33,1 (29,) | 28,3 (29,8) | 17,9 (22,9) | 21,7 (26,9) |
| Comorbilidades (Charlson) | 0,9 (1,2) | 0,9 (1,1) | 0,9 (1,2) | 0,8 (1,3) | 1,1 (1,2) |
| Índice de masa corporal (kg/m2) | 27,1 (5,3) | 27,6 (5,4) | 27,2 (5,1) | 26,3 (4,8) | 26,3 (6,5) |
| FEV1 (%) | 76,6 (26,8) | 75,5 (26,) | 77,1 (25,8) | 80,2 (28,7) | 68, (30,8) |
| Alfa-1 antitripsina (mg/dl) | 100,1 (27,2) | 118,5 (18,9) | 109,0 (13,8) | 79,0 (8,0) | 35,5 (16,9) |
Datos expresados con media (desviación estándar) o frecuencias absolutas (relativas) según la naturaleza de las variables.
AAT: alfa-1 antitripsina; FEV1: volumen espirado forzado en un segundo; PCR: proteína C reactiva.
El presente estudio pone de manifiesto y cuantifica el impacto en la detección de casos de DAAT con un programa de detección de casos sistemático en pacientes con EPOC. Nuestros resultados indican que: 1)los casos de DAAT existen en la comunidad; 2)es necesario tener una actitud proactiva en la detección de los mismos mediante un programa de detección de casos sistemático; 3)el criterio clínico no es suficiente para detectar casos de DAAT, y 4)los criterios para la selección de casos para estudio genético modifican el resultado del estudio sistemático.
El DAAT es una condición clínica que, en sus formas más graves, condiciona la aparición de enfermedad pulmonar y hepática. Sin embargo, su manifestación clínica final está matizada, al menos, por dos factores clave. Por un lado, la herencia codominante del trastorno, que hace que ambos alelos se expresen. Esto hace que no exista una condición absoluta de sano/enfermo, sino que la concentración hemática de AAT depende de los dos alelos que tenga cada sujeto afecto de manera individual. Este límite de la concentración hemática se ha establecido en 11μM18. Por otro lado, la importante influencia de otros factores de riesgo, como la exposición a tóxicos inhalados como parte de la patogenia de la afectación respiratoria, influye de manera decisiva en el grado de afectación respiratoria19. Estos dos factores, y otros de índole más biológica, hacen que la penetrancia del trastorno sea variable. Consecuentemente, en la actualidad no existe ninguna variable clínica, funcional o radiológica que permita sospechar por sí sola con un alto grado de certeza la presencia de este déficit. Por tanto, la única forma que actualmente tenemos de identificar casos es mediante un programa de detección de casos sistemático de los pacientes con EPOC. Adicionalmente, se ha propuesto que sería necesario cribar otras enfermedades de la vía aérea, como las bronquiectasias20, y se está estudiando su impacto en otras broncopatías, como el asma grave21 o la fibrosis quística22. Sin embargo, a pesar de un aumento en la frecuencia de las pruebas diagnósticas para el DAAT descritas en el Reino Unido en las últimas décadas, solo el 2,2% de los pacientes diagnosticados con EPOC antes de los 60años son estudiados2.
Diversos países han establecido programas de detección de casos en población adulta con resultados notables. En Italia, un estudio reciente evaluó la frecuencia de alelos relacionados con el DAAT en 200 individuos con EPOC empleando el mismo punto de corte que nosotros para el genotipado7. Tras genotipar a 141 pacientes, los autores encontraron un total de 36 pacientes que tenían una deficiencia no grave. Además, 5pacientes tenían un DAAT grave, lo que establece una detección del 29,1% considerando todas las combinaciones alélicas, una cifra inferior a la nuestra. En Brasil, un estudio evaluó la frecuencia de los alelos relacionados con el DAAT en 551 casos con EPOC6. Entre estos, 40 (7,2%) tenían alguna mutación genética, mientras que 11 (2%) tenían un genotipo Pi*ZZ, lo que resultó en una enfermedad respiratoria grave. En el Reino Unido, la prevalencia del DAAT fue del 23,7% en los pacientes con EPOC que fueron evaluados2. En Portugal, evaluaron una muestra de 1.684 individuos entre 2006 y 2015, en base a un análisis retrospectivo de la base de datos de un laboratorio que ofrece el servicio de diagnóstico genético de DAAT5. Este estudio identificó 417 sujetos (24,7%) con DAAT grave y 761 portadores (45,2%). En Alemania, entre 2003 y 2015 se analizaron los resultados de más de 18.000 pruebas diagnósticas por un laboratorio de referencia8, de los que el 37,12% portaban al menos una mutación. De ellos, los autores identificaron un 9,82% de pacientes con DAAT grave. En Polonia, en 2010 se inició el programa de detección de DAAT dirigido a pacientes con trastornos respiratorios. Después de 4años, se examinaron casi 2.500 pacientes con trastornos pulmonares obstructivos crónicos y, en esta cohorte, el 13% tenían alelos relacionados con el DAAT9. En Irlanda, el programa nacional irlandés de detección dirigida identificó un 27,1% de portadores de algún alelo relacionado con el DAAT sobre una muestra de 3.000 individuos23. En Estados Unidos, entre 2007 y 2009 se evaluaron un total de 3.457 pacientes, de los cuales 3.152 fueron elegibles24. Los pacientes deficitarios (ZZ, SZ) constituían el 0,63% de los sujetos, mientras que el 10,88% eran portadores de otras combinaciones alélicas (MS, MZ). En España, el principal estudio disponible se publicó en el año 2005 y empleaba una metodología similar a la de nuestro segundo periodo, con el análisis de los alelos S y Z en muestras de gota seca de pacientes con disminución de proteína en sangre25. Los autores encontraron un total de 8 (0,37%) individuos con la deficiencia severa de PI*ZZ y 3pacientes con el genotipo PI*SZ (0,3).
El análisis de los resultados de estos estudios previos y los del presente estudio nos dejan dos reflexiones importantes. La primera se deriva de estudios previos que indican que la península ibérica tiene una prevalencia más elevada de alelos relacionados con el DAAT, especialmente S y Z, que otras áreas geográficas26,27. Estos datos justifican y hacen especialmente necesario que todos los centros que atiendan a pacientes con EPOC tengan establecido un programa de detección de casos adecuado. La segunda reflexión es sobre el punto de corte de la concentración hemática de AAT para realizar un estudio genético. A pesar de que un DAAT debe implicar una disminución de esta concentración hemática, se ha descrito que pacientes portadores con concentraciones hemáticas dentro del rango de normalidad pueden ser portadores de algún alelo relacionado con el DAAT. Consecuentemente, los programas de detección de casos han optado por elevar el punto de corte para solicitar un estudio genético. Estos puntos de corte varían según el documento consulado: ≤110mg/dl según la normativa portuguesa y europea28,29 basada en un estudio de laboratorio30, 90mg/dl según el documento español31, recientemente actualizado a un punto de corte de 116mg/dl32, 120mg/dl en Italia33, 100mg/dl en Irlanda34, o <116mg/dl, recientemente propuesto por la Sociedad Española de Médicos de Atención Primaria35.
Otro debate abierto se refiere a si realmente es necesario detectar a portadores sanos, con el aumento de consumo de recursos que conlleva, o si limitarse a detectar casos que realmente pueden tener un impacto clínico relevante partiendo de una concentración inicial <90mg/dl. Las diferencias entre ambas estrategias quedan claramente puestas de manifiesto en nuestro estudio si se comparan los casos detectados en los periodos 2 y3. Aunque ambos son claramente superiores al periodo1, cuando no se hacía detección de casos sistemático, en el periodo3, con un punto de corte más elevado y con la capacidad de detectar mayor número de mutaciones, los casos identificados son mayores en número. La actitud en nuestra unidad es optar por detectar también los portadores sanos. Esto es debido a que esta detección permite hacer un cribado familiar mayor que posibilite la identificación de casos graves en edades tempranas en los que hacer una intervención preventiva.
Lo que sí parece claro, tanto por nuestros resultados como por los resultados de estudios previos comentados más arriba, es que la sospecha clínica es un mal criterio para identificar casos. En nuestro periodo1, en el que solo se estudiaban las sospechas clínicas, apenas se habían detectados casos, y la mayoría habían sido detectados en otros servicios o centros. Además, durante los periodos 2 y3 los escasos casos diagnosticados por sospecha clínica no eran graves. Estos datos apoyan la idea de que no existe una característica clínica, radiológica o funcional que lleve a la identificación de los casos con DAAT y refuerza la idea de la necesidad de hacer un programa de detección de casos sistemático en pacientes con EPOC.
Nuestro estudio tiene la fortaleza de poder describir los cambios en la detección de casos con DAAT en tres periodos consecutivos con políticas de detección de casos diferentes, lo que permite comparar la rentabilidad diagnóstica de cada protocolo. Además, la aplicación sistemática en una consulta monográfica de EPOC permite estimar el número de casos que podrían detectarse en una consulta similar con una población de referencia acorde. Sin embargo, para poder interpretar bien los resultados es necesario tener presente algunas consideraciones metodológicas. Es importante tener presente que se trata de una población de sujetos con enfermedad respiratoria. Sin embargo, el DAAT puede afectar también al hígado. Es probable que el número de casos detectados aumentase si se incluyera en el análisis la misma detección sistemática de casos para pacientes con hepatopatías. Otro aspecto a considerar es que el estudio de las mutaciones se hace por genotipado directo, explorando la presencia de mutaciones concretas. Esto implica que el diagnóstico de genotipo normal PI*MM se hace por exclusión. Por este motivo, es importante tener los valores de la concentración hemática de AAT y, en caso de presentar valores no normales y un genotipo PI*MM, proceder a la secuenciación del gen.
En conclusión, el presente estudio describe los cambios en la detección de pacientes portadores de alelos relacionados con el DAAT a lo largo del tiempo, con tres periodos en los que se aplican políticas de identificación de casos diferentes. Los datos claramente avalan la utilización de un sistema de detección de casos sistemático en la población de pacientes con EPOC como mejor forma de identificar casos con DAAT. Consecuentemente, todos los centros que atiendan a pacientes con EPOC deberían tener diseñado e implementado un sistema de detección de casos de DAAT para poder identificarlos lo antes posible, antes de que el deterioro pulmonar esté en una fase avanzada.
FinanciaciónEl estudio de los genotipos del DAAT está financiado por Grifols SA. La herramienta REDCap está financiada por la Sociedad Española de Neumología y Cirugía Torácica (SEPAR).
Contribuciones de los autoresJLLC ha diseñado el estudio y coordinado la puesta a punto del programa de detección de casos, la realización del análisis de la base de datos y la elaboración del borrador original.
RRA ha ayudado en la coordinación del estudio, la inclusión de casos y la revisión del manuscrito final, así como en la decisión de publicar.
RRS y DNO han realizado parte del trabajo de campo en la implementación del programa de cribado y han participado en la revisión del manuscrito final, así como en la decisión de publicar.
BRD, FOR, EMM y LCH han colaborado en la inclusión de casos y en la revisión del manuscrito final, así como en la decisión de publicar.
Conflictos de interésCP, JLLC ha recibido honorarios en los últimos tres años por impartir conferencias, asesoría científica, participación en estudios clínicos o redacción de publicaciones para (en orden alfabético): AstraZeneca, Bial, Boehringer, Chiesi, CSL Behring, Faes, Gebro, Grifols, GSK, Menarini. El resto de los autores no tienen conflicto de intereses.
