



There is little information on inflammatory myopathies in Colombia. The objective was to identify the demographic and clinical characteristics of these patients in two tertiary care hospitals between 2010 and 2015.
Materials and methodsA descriptive, retrospective survey was carried out, by reviewing medical records and obtaining information on demographic and clinical variables. The qualitative variables were expressed using absolute and relative frequencies, and the quantitative with mean and standard deviation (SD), or median with interquartile ranges (IQR), depending on data distribution. The IBM SPSS 22 statistical package was used.
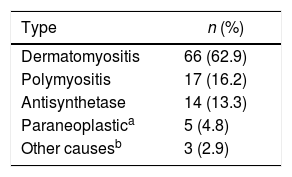
ResultsA total of 105 patients with a mean age of 50.4 years (SD: 15.1) were included, with 76 (72.4%) women. In total, 50 subjects (48.5%) had a definitive diagnosis. The most common inflammatory myopathy was dermatomyositis (n = 66; 62.9%). The skin was the most commonly affected organ (n=66; 62.9%). Muscle weakness was present in 60 individuals (57.1%). The most frequent alarm sign was swallowing disorder (n = 28; 26.7%). Creatine phosphokinase was higher in polymyositis, with a median of 1800 IU/L (IQR: 365–6157). The most widely used drugs were glucocorticoids (n = 83; 79%). Some patients were refractory to immunosuppressive treatment, mainly in antisynthetase syndrome (n = 5; 35.7%). Five patients (4.8%) died of infections (pneumonia and bacteraemia).
ConclusionsIn this cohort, the most common entity was dermatomyositis, and the most affected organ was the skin. There was a significant presentation of warning signs, refractoriness to immunosuppressive treatment, and lower muscle enzyme values compared to other cohorts. Mortality was mainly due to infectious complications.
Existe poca información sobre las miopatías inflamatorias en Colombia. El objetivo fue identificar las características demográficas y clínicas de estos pacientes en dos instituciones de alta complejidad entre los años 2010 y 2015.
Materiales y métodosSe realizó un estudio descriptivo y retrospectivo. Mediante revisión de registros médicos, se obtuvo información sobre variables demográficas y clínicas. Las variables cualitativas se expresaron mediante frecuencias absolutas y relativas, y las cuantitativas con media y desviación estándar (DE) o mediana con rangos intercuartílicos (RIQ), dependiendo de la distribución de los datos. Se utilizó el paquete estadístico IBM SPSS® v.22.
ResultadosSe incluyeron 105 pacientes con edad promedio de 50,4 años (DE: 15,1); 76 mujeres (72,4%). En total, 50 sujetos (48,5%) tuvieron diagnóstico definitivo. La miopatía inflamatoria más común fue dermatomiositis (n = 66; 62,9%). La piel fue el órgano más comúnmente afectado (n = 66; 62,9%). La debilidad muscular estuvo presente en 60 individuos (57,1%). El signo de alarma más frecuente fue el trastorno de la deglución (n = 28; 26,7%). La creatinfosfoquinasa tuvo mayor elevación en polimiositis con una mediana de 1.800 UI/l (RIQ: 365–6.157). Los medicamentos más utilizados fueron los glucocorticoides (n = 83; 79%). Hubo refractariedad al tratamiento inmunosupresor, principalmente en síndrome antisintetasa (n = 5; 35,7%). Cinco pacientes (4,8%) murieron por infecciones (neumonía y bacteriemia).
ConclusionesEn esta cohorte, la entidad más común fue la dermatomiositis y el órgano más afectado fue la piel. Hubo presentación relevante de signos de alarma, refractariedad al tratamiento inmunosupresor y valores de enzimas musculares menores comparados con otras cohortes. La mortalidad fue principalmente por complicaciones infecciosas.
Inflammatory myopathies (IIMs) are a heterogeneous group of acquired chronic autoimmune diseases, with multisystem involvement,1 which have an unknown etiology, autoimmune pathophysiology and their prognosis is variable. The IIMs include dermatomyositis (DM), polymyositis (PM), inclusion body myopathies (IBM), necrotizing myositis (NM), inflammatory myopathy – as part of an overlap syndrome with other autoimmune diseases –, paraneoplastic syndrome and antisynthetase syndrome (SAS).2–6
In the IIMs, the onset of symptoms, which is usually acute or subacute, is manifested mainly with proximal and symmetrical muscle weakness, secondary to inflammation and necrosis of the muscle fiber.3,7,8 As the disease progresses, chronic and irreversible atrophy can develop, which is a major cause of disability and morbidity.3,7
As systemic diseases, IIMs can be associated with extramuscular manifestations, among which the most frequent are cutaneous and pulmonary. One of the latter is the antisynthetase syndrome, which entails a worse prognosis.3,7,9
The clinical course of the IIMs is variable, with monocyclic, polycyclic and persistently active patterns.10 The treatment of these entities is based on glucocorticoids, but other immunomodulators and immunosuppressants can also be used.7,10
Accurately estimating epidemiological data on IIMs is difficult, given their low prevalence and the variations in classification criteria.8 Systematic reviews and cohort studies have calculated a global incidence that ranges between 6 patients/1,000,000/year and a prevalence of 14/100,000 inhabitants.11–13 In the EuroMyositis registry13 DM was found to be the most common subtype of IIM (31%). In adults, these diseases occur between the ages of 45 and 65 years,14,15 more frequently in women, with a female to male ratio (2:1).13,15–17
There are few studies In Latin America regarding the epidemiology and behavior of these diseases,18 and there are differences with what has been published in the global literature.
Some case reports19–22 and descriptive studies23,24 about these entities had been published in Colombia. In recent years, their classification, conception and treatment have changed, and for this reason it is relevant and necessary to obtain updated information on these diseases. The objective of the present study was to describe the demographic and clinical characteristics of the patients with IIM in 2 institutions of high complexity.
Materials and methodsStudy design and selection of patientsAn observational, descriptive, retrospective study of a cohort of patients with IIM admitted to 2 centers of high complexity between the years 2010 and 2015 was conducted.
Patients over 18 years of age, with a diagnosis of DM or PM established by a rheumatologist or who met the modified Bohan and Peter criteria were included.6,25–28 Patients with a diagnosis of another rheumatological disease were excluded.
Information collection processThe information of the study was obtained by reviewing the physical and electronic medical records of the patients who met the inclusion criteria; the data were recorded in an electronic form designed with the MAGPI tool, according to the variables that were necessary to meet the objectives of the study. A pilot test was carried out with 12 medical records, in order to standardize the collection process, verify the quality of the recorded data and make possible adjustments to the form.
In the case of the variables that were not found in the selected medical history records, a search was carried out in nursing records, laboratory results, and assessments made by other specialties.
The variables collected were the following:
- •
Demographic: age, sex, date of birth and age in years at the time of diagnosis.
- •
Anthropometric: weight, height and body mass index.
- •
Clinical characteristics: muscle involvement (distal or proximal), muscle strength scale (according to the Medical Research Council)29 and comorbidities (neoplasms, infectious, cardiovascular, systemic).
- •
The IMMs were classified according to the modified Bohan and Peter criteria.30 The European Neuromuscular Center (ENMC) Criteria 2011 were taken into account for the inclusion body miopathy,31 while in the case of necrotizing myopathy, the clinical findings and the temporal relationship with the use of medications were taken into consideration.32
- •
Clinical outcomes: hospitalization, admission to intensive care unit, death, recurrent infections (defined as more than one episode of infection in the last six months that would have required hospital management), complications, functional limitation determined by the Steinbroker functional class,33 deterioration of the renal function (increase greater than 0.3 mg/dl/in 48 h or more than 1.5-fold the baseline creatinine value),34 refractoriness to treatment (defined as failure to achieve remission of the disease after a dose of 0.5 mg/kg of methylprednisolone during one month, with progressive dismount in the next three months or inability to obtain improvement after treating with second-line immunosuppressive therapy or immunoglobulin).35
- •
Laboratory: total creatine phosphokinase (CPK), lactate dehydrogenase (LDH), aldolase, transaminases (AST, ALT), autoimmunity tests (ANA, anti-ENA, anti Jo-1).
- •
Diagnostic aids: magnetic resonance imaging (MRI), electromyography (EMG) and muscle biopsy.
- •
Treatment: glucocorticoids and other immunosuppressive and immunomodulatory drugs (chloroquine, hydroxychloroquine, methotrexate, mycophenolate mofetil, intravenous immunoglobulin, cyclosporine, tacrolimus, azathioprine, and rituximab) according to the type of inflammatory myopathy.
The information was exported to a Microsoft Excel® 2011 database, which contained fields with data entry restrictions in order to reduce possible typing errors; in addition, a categorization of the quantitative variables was made according to the clinical criteria. Before proceeding with the analysis of the information, its consistency was verified by the exploration of the values and the concordance of the recorded data. In case of any confusing data, a new review of the medical history was made.
Analysis planThe qualitative variables were expressed in absolute and relative frequencies, while the mean and the standard deviation (SD), or the median with their respective interquartile ranges were used for the quantitative variables, depending on the distribution of the data. The statistical analyses were carried out with the IBM SPSS® v.22 statistic package.
Control of biasSelection biasesThey were controlled by a rigorous and exhaustive review of the medical records of the patients who met the eligibility criteria for the study.
Information biasesIn order to control for possible information biases, a pilot test was carried out, meetings on the collection process were held, and a database was designed using fields with entry restrictions.
Ethical considerationsAccording to Resolution 8430 of 1993, article 11, of the Colombian Ministry of Health, this research is considered without risk, since retrospective documentary methods were used, conducting a review of medical records, with prior approval of the research ethics committee of the participating institutions.
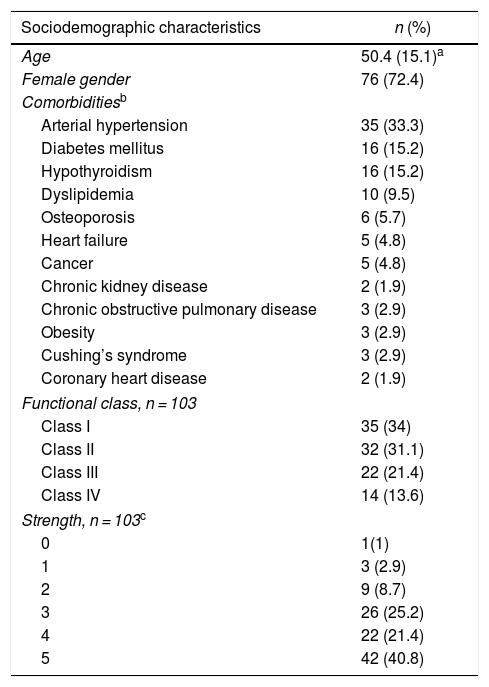
ResultsDemographic characteristics and comorbidities315 clinical records were reviewed, of which 105 met eligibility criteria: 76 patients (72.4%) were women; the mean age was 50.4 (±15.1) years at the time of entering the cohort. The most frequent comorbidity was arterial hypertension (n = 35; 33.3%), followed by diabetes mellitus (n = 16; 15.2%) and hypothyroidism (n = 16; 15.2%). As for the functional class at the time of entering the cohort, the majority had classes II (31.1%) and III (21.4%). Fifty one subjects (48.6%) required hospitalization and 7 (6.7%) were admitted to the intensive care unit (Table 1).
Sociodemographic and clinical characteristics of the patients with inflammatory myopathy.
| Sociodemographic characteristics | n (%) |
|---|---|
| Age | 50.4 (15.1)a |
| Female gender | 76 (72.4) |
| Comorbiditiesb | |
| Arterial hypertension | 35 (33.3) |
| Diabetes mellitus | 16 (15.2) |
| Hypothyroidism | 16 (15.2) |
| Dyslipidemia | 10 (9.5) |
| Osteoporosis | 6 (5.7) |
| Heart failure | 5 (4.8) |
| Cancer | 5 (4.8) |
| Chronic kidney disease | 2 (1.9) |
| Chronic obstructive pulmonary disease | 3 (2.9) |
| Obesity | 3 (2.9) |
| Cushing’s syndrome | 3 (2.9) |
| Coronary heart disease | 2 (1.9) |
| Functional class, n = 103 | |
| Class I | 35 (34) |
| Class II | 32 (31.1) |
| Class III | 22 (21.4) |
| Class IV | 14 (13.6) |
| Strength, n = 103c | |
| 0 | 1(1) |
| 1 | 3 (2.9) |
| 2 | 9 (8.7) |
| 3 | 26 (25.2) |
| 4 | 22 (21.4) |
| 5 | 42 (40.8) |
The inflammatory myopathy was classified as definitive in 50 individuals (48.5%). In addition, 60 patients (57.1%) presented symmetric muscle weakness, followed in frequency by swallowing disorders (26.7%).
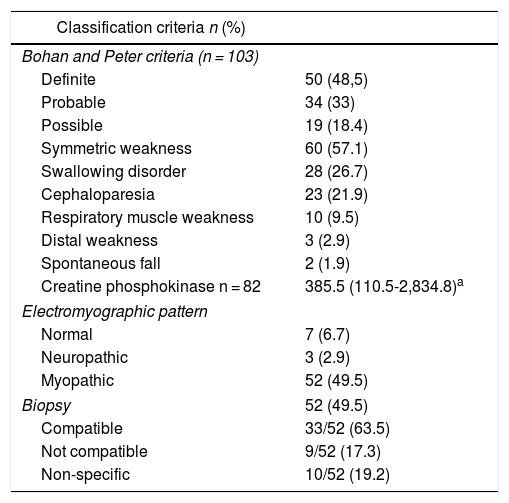
Regarding the diagnostic aids, a muscle MRI was performed in 44 subjects (41.9%), of whom 8 (18.2%) had a result compatible with inflammatory myopathy (Table 2). Dermatomyositis was the most frequent type of inflammatory myopathy (n = 66; 62.9%) (Table 3).
Classification criteria, predictors of severity and diagnostic aids in an inflammatory myopathy cohort.
| Classification criteria n (%) | |
|---|---|
| Bohan and Peter criteria (n = 103) | |
| Definite | 50 (48,5) |
| Probable | 34 (33) |
| Possible | 19 (18.4) |
| Symmetric weakness | 60 (57.1) |
| Swallowing disorder | 28 (26.7) |
| Cephaloparesia | 23 (21.9) |
| Respiratory muscle weakness | 10 (9.5) |
| Distal weakness | 3 (2.9) |
| Spontaneous fall | 2 (1.9) |
| Creatine phosphokinase n = 82 | 385.5 (110.5-2,834.8)a |
| Electromyographic pattern | |
| Normal | 7 (6.7) |
| Neuropathic | 3 (2.9) |
| Myopathic | 52 (49.5) |
| Biopsy | 52 (49.5) |
| Compatible | 33/52 (63.5) |
| Not compatible | 9/52 (17.3) |
| Non-specific | 10/52 (19.2) |
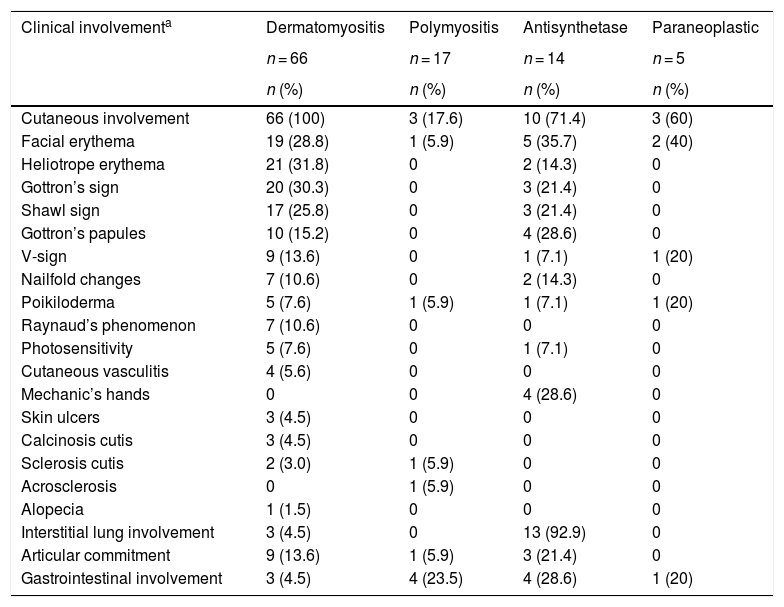
The skin was the organ most frequently involved, especially in dermatomyositis (n = 66; 100%), and of these cutaneous manifestations, heliotrope erythema (n = 21; 31.8%) and Gottron’s sign (n = 20; 30.3%) were the most frequent.
In the antisynthetase syndrome the lung was the most affected organ (n = 13; 92.9%), mainly due to interstitial lung disease, while articular commitment was present in 3 (21.4%) patients (Table 4).
Most frequent clinical characteristics according to the type of myopathy.
| Clinical involvementa | Dermatomyositis | Polymyositis | Antisynthetase | Paraneoplastic |
|---|---|---|---|---|
| n = 66 | n = 17 | n = 14 | n = 5 | |
| n (%) | n (%) | n (%) | n (%) | |
| Cutaneous involvement | 66 (100) | 3 (17.6) | 10 (71.4) | 3 (60) |
| Facial erythema | 19 (28.8) | 1 (5.9) | 5 (35.7) | 2 (40) |
| Heliotrope erythema | 21 (31.8) | 0 | 2 (14.3) | 0 |
| Gottron’s sign | 20 (30.3) | 0 | 3 (21.4) | 0 |
| Shawl sign | 17 (25.8) | 0 | 3 (21.4) | 0 |
| Gottron’s papules | 10 (15.2) | 0 | 4 (28.6) | 0 |
| V-sign | 9 (13.6) | 0 | 1 (7.1) | 1 (20) |
| Nailfold changes | 7 (10.6) | 0 | 2 (14.3) | 0 |
| Poikiloderma | 5 (7.6) | 1 (5.9) | 1 (7.1) | 1 (20) |
| Raynaud’s phenomenon | 7 (10.6) | 0 | 0 | 0 |
| Photosensitivity | 5 (7.6) | 0 | 1 (7.1) | 0 |
| Cutaneous vasculitis | 4 (5.6) | 0 | 0 | 0 |
| Mechanic’s hands | 0 | 0 | 4 (28.6) | 0 |
| Skin ulcers | 3 (4.5) | 0 | 0 | 0 |
| Calcinosis cutis | 3 (4.5) | 0 | 0 | 0 |
| Sclerosis cutis | 2 (3.0) | 1 (5.9) | 0 | 0 |
| Acrosclerosis | 0 | 1 (5.9) | 0 | 0 |
| Alopecia | 1 (1.5) | 0 | 0 | 0 |
| Interstitial lung involvement | 3 (4.5) | 0 | 13 (92.9) | 0 |
| Articular commitment | 9 (13.6) | 1 (5.9) | 3 (21.4) | 0 |
| Gastrointestinal involvement | 3 (4.5) | 4 (23.5) | 4 (28.6) | 1 (20) |
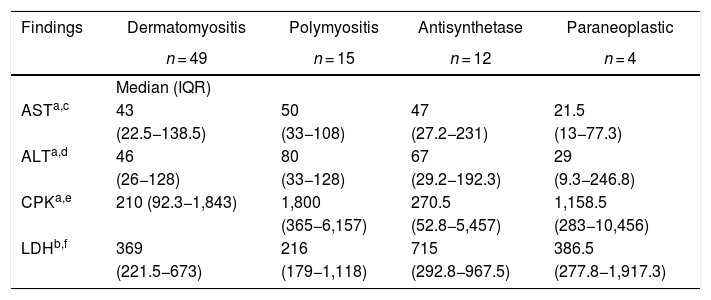
The muscle enzymes with the highest elevation in all groups of myopathies were total CPK and LDH. The total CPK had a higher elevation in polymyositis, with a median of 1,800 IU/l (365−6.157), while for LDH a higher elevation was registered in the antisynthetase syndrome, with a median of 715 IU/l (292.8−967.5). Anti-Ro antibodies were positive in 50% of the patients with antisynthetase syndrome and in 11.1% of the individuals with polymyositis. Other results are shown in Table 5.
Laboratory findings by type of myopathy.
| Findings | Dermatomyositis | Polymyositis | Antisynthetase | Paraneoplastic |
|---|---|---|---|---|
| n = 49 | n = 15 | n = 12 | n = 4 | |
| Median (IQR) | ||||
| ASTa,c | 43 | 50 | 47 | 21.5 |
| (22.5−138.5) | (33−108) | (27.2−231) | (13−77.3) | |
| ALTa,d | 46 | 80 | 67 | 29 |
| (26−128) | (33−128) | (29.2−192.3) | (9.3−246.8) | |
| CPKa,e | 210 (92.3−1,843) | 1,800 | 270.5 | 1,158.5 |
| (365−6,157) | (52.8−5,457) | (283−10,456) | ||
| LDHb,f | 369 | 216 | 715 | 386.5 |
| (221.5−673) | (179−1,118) | (292.8−967.5) | (277.8−1,917.3) |
| n/N (%) | n/N (%) | n/N (%) | n/N (%) | |
|---|---|---|---|---|
| ANA | 38/46 (82.6) | 8/11 (72.7) | 5/9 (55.6) | 0 |
| ANA pattern | ||||
| Speckled | 23/30 (76.7) | 2/8 (25) | 3/5 (60) | |
| Homogeneous | 4/30 (13.3) | 6/8 (75) | 1/5 (20) | |
| Nucleolar | 1/30 (3.3) | 0 | 1/5 (20) | |
| Cytoplasmic | 2/30 (6.7) | 0 | 0 | |
| Positive anti-Jo1 | 0 | 0 | 4/8 (50) | 0 |
| Positive anti-Ro | 3/35 (8.6) | 1/9 (11.1) | 5/10 (50) | 0 |
ANA: antinuclear antibodies; ALT: alanine aminotransferase; AST: aspartate aminotransferase; CPK: total creatine phosphokinase; LDH: lactate dehydrogenase; IQR: interquartile ranges.
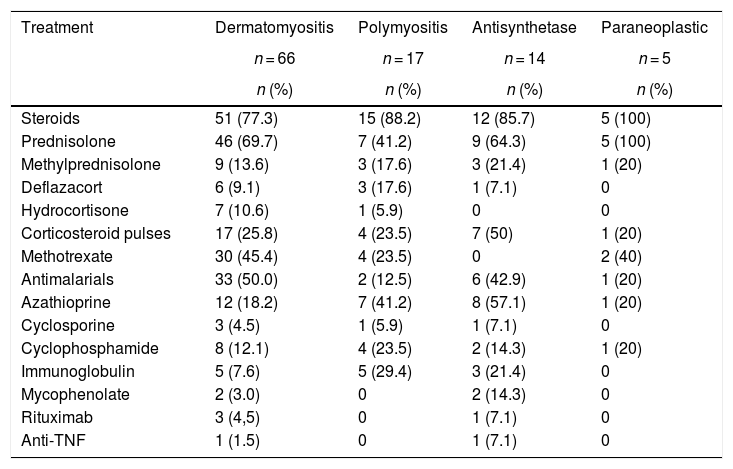
Corticosteroids, used in 83 (81.3%) subjects, were the main treatment in all types of myopathies. The corticosteroid most frequently used was prednisolone (n = 67; 65.7%), followed by methylprednisolone (n = 16; 15.7%). Seven (50%) individuals with antisynthetase syndrome required pulses of corticosteroids, while in the group of paraneoplastic myopathies only one required this therapy.
On the other hand, antimalarials were used with higher frequency in patients with dermatomyositis (n = 33; 50%); azathioprine (AZA) was used in 8 (57.1%) of the patients with antisynthetase syndrome. Finally, mycophenolate mofetil (MF) was used in 2 patients with antisynthetase syndrome. Methotrexate (MTX) was used in 36 (33.3%) patients with all types of myopathy. The rest of the immunosuppressive management is described in Table 6. Two patients received anti-TNF for refractory inflammatory myopathy: one female patient with dermatomyositis was treated with adalimumab and one patient with antisynthetase syndrome was treated with etanercept.
Treatment according to the type of myopathy.
| Treatment | Dermatomyositis | Polymyositis | Antisynthetase | Paraneoplastic |
|---|---|---|---|---|
| n = 66 | n = 17 | n = 14 | n = 5 | |
| n (%) | n (%) | n (%) | n (%) | |
| Steroids | 51 (77.3) | 15 (88.2) | 12 (85.7) | 5 (100) |
| Prednisolone | 46 (69.7) | 7 (41.2) | 9 (64.3) | 5 (100) |
| Methylprednisolone | 9 (13.6) | 3 (17.6) | 3 (21.4) | 1 (20) |
| Deflazacort | 6 (9.1) | 3 (17.6) | 1 (7.1) | 0 |
| Hydrocortisone | 7 (10.6) | 1 (5.9) | 0 | 0 |
| Corticosteroid pulses | 17 (25.8) | 4 (23.5) | 7 (50) | 1 (20) |
| Methotrexate | 30 (45.4) | 4 (23.5) | 0 | 2 (40) |
| Antimalarials | 33 (50.0) | 2 (12.5) | 6 (42.9) | 1 (20) |
| Azathioprine | 12 (18.2) | 7 (41.2) | 8 (57.1) | 1 (20) |
| Cyclosporine | 3 (4.5) | 1 (5.9) | 1 (7.1) | 0 |
| Cyclophosphamide | 8 (12.1) | 4 (23.5) | 2 (14.3) | 1 (20) |
| Immunoglobulin | 5 (7.6) | 5 (29.4) | 3 (21.4) | 0 |
| Mycophenolate | 2 (3.0) | 0 | 2 (14.3) | 0 |
| Rituximab | 3 (4,5) | 0 | 1 (7.1) | 0 |
| Anti-TNF | 1 (1.5) | 0 | 1 (7.1) | 0 |
TNF: tumor necrosis factor.
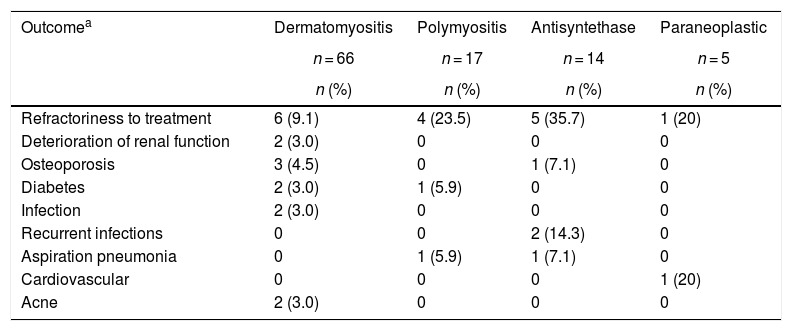
The most frequent complication was refractoriness to immunosuppressive treatment, mainly in antisynthetase syndrome (n = 5; 35.7%) and in polymyositis (n = 4; 23.5%); the second in frequency corresponded to recurrent infections in the group of antisynthetase syndrome (n = 2; 14.3%), followed by aspiration pneumonia in antisynthetase syndrome (n = 1; 7.1%) and in polymyositis (n = 1; 5.9%) (Table 7). Two patients had renal involvement, which was explained by etiologies other than inflammatory myopathies.
Clinical outcomes according to the type of myopathy.
| Outcomea | Dermatomyositis | Polymyositis | Antisyntethase | Paraneoplastic |
|---|---|---|---|---|
| n = 66 | n = 17 | n = 14 | n = 5 | |
| n (%) | n (%) | n (%) | n (%) | |
| Refractoriness to treatment | 6 (9.1) | 4 (23.5) | 5 (35.7) | 1 (20) |
| Deterioration of renal function | 2 (3.0) | 0 | 0 | 0 |
| Osteoporosis | 3 (4.5) | 0 | 1 (7.1) | 0 |
| Diabetes | 2 (3.0) | 1 (5.9) | 0 | 0 |
| Infection | 2 (3.0) | 0 | 0 | 0 |
| Recurrent infections | 0 | 0 | 2 (14.3) | 0 |
| Aspiration pneumonia | 0 | 1 (5.9) | 1 (7.1) | 0 |
| Cardiovascular | 0 | 0 | 0 | 1 (20) |
| Acne | 2 (3.0) | 0 | 0 | 0 |
Five patients (4,8%) died during the first assessment, of them, 2 had paraneoplastic myopathy, one had dermatomyositis, one had polymyositis and another had antisynthetase syndrome; 4 of these patients died from infectious complications such as pneumonia and bacteremia, while one patient died from cardiorespiratory arrest secondary to respiratory failure.
DiscussionAs far as is known, this is the largest study on inflammatory myopathies in Colombia.
It was found a higher proportion of affected women, which is consistent with other local and international studies.12,13,15,23,36
Within the comorbidities, the findings are similar to those of other series of patients.37–39
On the other hand, the significant number of patients who required admission to the hospital and to the intensive care unit stands out, which is an indication of a chronic, progressive and serious course. Other authors have also described a higher rate of hospitalization in patients with inflammatory myopathies,40,41 which is related to the progressive course of the disease.40
There are currently several classification criteria for inflammatory myopathies (Bohan and Peter,27,28 Tanimoto et al.,42 Targoff et al.,43 Dalakas and Hohlfeld,2 ENMC30), however, most of them have not been widely validated. The most widely used in the different clinical studies are those of Bohan and Peter, with high sensitivity (98%) and low specificity (55%).27,28 In our study, the diagnosis of inflammatory myopathy was established by the clinical criteria of the treating rheumatologist, while the patients were classified according to the modified Bohan and Peter criteria. Recently, the new classification criteria of the American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) have been published, which have demonstrated a good correlation with the Bohan and Peter criteria, widely used, with a sensitivity of 93% and a specificity of 88% when the information of the biopsy is available.44 The foregoing shows that the classification criteria used in the cohort have an adequate performance, even when compared with the new criteria.
The muscle involvement in the majority of the patients was given by proximal and symmetric weakness. In the study conducted by Pinto et al.,23 muscle weakness in the pelvic girdle was found in up to 80% of the patients, and cutaneous findings were present in dermatomyositis, similar to those described in our cohort, except for a higher frequency of Raynaud’s phenomenon, vasculitic changes, mechanic’s hands, poikiloderma and calcinosis cutis.
Other extramuscular manifestations such as dysphagia, articular involvement and diffuse interstitial lung disease (DILD) were similar to the findings of the study of Pinto et al.23 On the other hand, in a study published by Dobloug et al., muscle weakness was found as the predominant finding in 84% of the patients, dyspnea in 44% of DM and 22% of PM, dysphagia in 23% and Raynaud’s phenomenon together with mechanic’s hands, which were more frequent in DM.45 When comparing what was previously described with the data reported by the EuroMyositis registry,13 it can be seen that there was similarity in muscle weakness as a cardinal symptom of the different subtypes of myopathies; however, in this registry there was a higher frequency of some cutaneous manifestations such as Raynaud phenomenon, mechanic’s hands, calcinosis cutis and skin ulcers; while in SAS the articular involvement was greater than that described in our cohort.
With regard to laboratory aids, it should be clarified that only some myositis-specific antibodies (anti-Jo1, and anti-Pm/Scl) are found; however, in the description of the ACR/EULAR classification criteria30 the lack of availability of data on specific antibodies was also evident.
When evaluating the laboratory results, it is striking that half of the patients with antisynthetase were positive for anti-Ro and anti-Jo1. Koenig et al.46 showed that anti-Jo1 antibodies were found concomitantly with anti-Ro in 73%, whereas a lower response to prednisolone and a greater requirement of second-line therapy was found among those patients with anti-Jo1 positivity, which could be an explanation of why in our cohort the antisynthetase subgroup was more refractory to treatment and required a greater amount of pulse of steroids..
In our series, the muscle enzymes that more elevation presented were total CPK and LDH; however, there are some differences with respect to other publications from Norway45 and Spain37 in which higher levels of total CPK were found in patients with dermatomyositis, polymyositis and paraneoplastic syndrome. Some hypotheses could explain this finding, such as the fact that many patients came with diagnosis and immunosuppressive treatment prior to entering to the study, and it is possible that this decreases the total CPK levels. Another explanation for these findings is that the patients with long-standing IIM can present with total CPK values close to normal, which occurs when the greatest part of the muscle has been affected and is replaced with fatty tissue. The low specificity of LDH in inflammatory myopathies should also be recognized.47
What is described in the present study is consistent with the treatment schemes analyzed by Meyer et al.48 Other studies, most of them retrospective cohorts,13,37,49 case series50 and some clinical trials,51–54 have described the use of steroids as a first line therapy in conjunction with other immunosuppresants.8,50 In a publication conducted by Nuño et al.,37 the treatment schemes used were similar to that found in this work. The use of anti-TNF in 2 patients from the cohort (one with dermatomyositis and the other with antisynthetase syndrome), in whom these drugs were used due to refractory disease, was striking. In none of the 2 cases there was a response; in addition, there is evidence of frequent relapses55 with their use as well as a possible induction and worsening of the inflammatory myopathy and the pulmonary involvement in these entities.56
The findings related to complications and mortality were similar to the infectious complications described in a study published by Murray et al.,57 in which it was reported that, of the 15,407 hospitalizations for DM and PM, 4.5% of the patients died and the main causes of mortality were pneumonia and infection. Infections in patients with IIM can be explained by the increased risk of bacterial and fungal infections, even in the absence of immunosuppressive management, which suggests defects in cell-mediated immunity that predispose to infections. Likewise, fewer deaths occurred regarding to other studies,23,49,58 since it is about the description of the entry of the patients to the cohort and there are no follow-up data over time.
The study has several limitations: selection bias was unavoidable, because the participating centers have a high level of complexity and it is possible that the patients would have higher severity of the disease or that they were widely studied. Additionally, there could be information biases associated with the retrospective study design, but we tried to control them with the strategies described in the materials and methods section.
The important number of patients included and the updated classification criteria stand out as strengths of this work.
ConclusionsA significant frequency of hospitalization and admission to the intensive care unit was found in a cohort of patients with inflammatory myopathies from Northwestern Colombia, which is related to a greater severity of the disease and refractoriness to treatment. Likewise, there were a significant number of patients with antisynthetase syndrome, who in turn presented greater refractoriness to treatment.
Conflict of interestThe authors declare that they have no conflict of interest.
Thanks to the participant institutions.
Please cite this article as: Salazar-Villa G, Rodríguez-Prada C, Bonfante-Tamara M, Restrepo-Correa R, Rodríguez-Padilla LM, Mesa-Navas MA, et al. Caracterización clínica de pacientes con miopatía inflamatoria en 2 instituciones de alta complejidad en Colombia: estudio descriptivo. Rev Colomb Reumatol. 2022;29:9–18.
