



Existe poca información sobre las miopatías inflamatorias en Colombia. El objetivo fue identificar las características demográficas y clínicas de estos pacientes en dos instituciones de alta complejidad entre los años 2010 y 2015.
Materiales y métodosSe realizó un estudio descriptivo y retrospectivo. Mediante revisión de registros médicos, se obtuvo información sobre variables demográficas y clínicas. Las variables cualitativas se expresaron mediante frecuencias absolutas y relativas, y las cuantitativas con media y desviación estándar (DE) o mediana con rangos intercuartílicos (RIQ), dependiendo de la distribución de los datos. Se utilizó el paquete estadístico IBM SPSS® v.22.
ResultadosSe incluyeron 105 pacientes con edad promedio de 50,4 años (DE: 15,1); 76 mujeres (72,4%). En total, 50 sujetos (48,5%) tuvieron diagnóstico definitivo. La miopatía inflamatoria más común fue dermatomiositis (n=66; 62,9%). La piel fue el órgano más comúnmente afectado (n=66; 62,9%). La debilidad muscular estuvo presente en 60 individuos (57,1%). El signo de alarma más frecuente fue el trastorno de la deglución (n=28; 26,7%). La creatinfosfoquinasa tuvo mayor elevación en polimiositis con una mediana de 1.800UI/l (RIQ: 365-6.157). Los medicamentos más utilizados fueron los glucocorticoides (n=83; 79%). Hubo refractariedad al tratamiento inmunosupresor, principalmente en síndrome antisintetasa (n=5; 35,7%). Cinco pacientes (4,8%) murieron por infecciones (neumonía y bacteriemia).
ConclusionesEn esta cohorte, la entidad más común fue la dermatomiositis y el órgano más afectado fue la piel. Hubo presentación relevante de signos de alarma, refractariedad al tratamiento inmunosupresor y valores de enzimas musculares menores comparados con otras cohortes. La mortalidad fue principalmente por complicaciones infecciosas.
There is little information on inflammatory myopathies in Colombia. The objective was to identify the demographic and clinical characteristics of these patients in two tertiary care hospitals between 2010 and 2015.
Materials and methodsA descriptive, retrospective survey was carried out, by reviewing medical records and obtaining information on demographic and clinical variables. The qualitative variables were expressed using absolute and relative frequencies, and the quantitative with mean and standard deviation (SD), or median with interquartile ranges (IQR), depending on data distribution. The IBM SPSS 22 statistical package was used.
ResultsA total of 105 patients with a mean age of 50.4 years (SD: 15.1) were included, with 76 (72.4%) women. In total, 50 subjects (48.5%) had a definitive diagnosis. The most common inflammatory myopathy was dermatomyositis (n=66; 62.9%). The skin was the most commonly affected organ (n=66; 62.9%). Muscle weakness was present in 60 individuals (57.1%). The most frequent alarm sign was swallowing disorder (n=28; 26.7%). Creatine phosphokinase was higher in polymyositis, with a median of 1800 IU/L (IQR: 365-6157). The most widely used drugs were glucocorticoids (n=83; 79%). Some patients were refractory to immunosuppressive treatment, mainly in antisynthetase syndrome (n=5; 35.7%). Five patients (4.8%) died of infections (pneumonia and bacteraemia).
ConclusionsIn this cohort, the most common entity was dermatomyositis, and the most affected organ was the skin. There was a significant presentation of warning signs, refractoriness to immunosuppressive treatment, and lower muscle enzyme values compared to other cohorts. Mortality was mainly due to infectious complications.
Las miopatías inflamatorias (MII) son un grupo heterogéneo de enfermedades autoinmunes crónicas adquiridas, con compromiso multisistemico1, que tienen una etiología desconocida, fisiopatología autoinmune y son de pronóstico variable. Entre las MII se encuentran: la dermatomiositis (DM), la polimiositis (PM), las miopatías por cuerpos de inclusión (MCI), la miositis necrosante (NM), la miopatía inflamatoria —como parte de un síndrome de superposición con otras enfermedades autoinmunes—, el síndrome paraneoplásico y el síndrome antisintetasa (SAS)2–6.
En las MII, el inicio de los síntomas, que suele ser agudo o subagudo, se manifiesta principalmente con debilidad muscular proximal y simétrica, secundaria a la inflamación y la necrosis de la fibra muscular3,7,8. Conforme progresa la enfermedad, puede generarse atrofia crónica e irreversible, que constituye una importante causa de discapacidad y morbilidad3,7.
Como enfermedades sistémicas, las MII pueden asociarse con manifestaciones extramusculares, entre las cuales las más frecuentes son las cutáneas y las pulmonares. Una de estas últimas es el síndrome antisintetasa, el cual conlleva un peor pronóstico3,7,9.
El curso clínico de las MII es variable, con patrones: monocíclico, policíclico y activos de forma persistente10. El tratamiento de estas entidades se basa en glucocorticoides, pero pueden utilizarse otros inmunomoduladores e inmunosupresores7,10.
Estimar con precisión los datos epidemiológicos de las MII es difícil, dada su baja prevalencia y las variaciones en los criterios de clasificación8. Revisiones sistemáticas y estudios de cohorte han calculado una incidencia global que oscila entre 6 pacientes/1.000.000/año y una prevalencia de 14/100.000 habitantes11–13. En el registro EuroMyositis13 se encontró la DM como el subtipo más común de las MII (31%). En los adultos estas enfermedades se presentan entre los 45 y los 65 años14,15, con más frecuencia en el género femenino, con una relación M:H (2:1)13,15–17.
En Latinoamérica existen pocos estudios con respecto a la epidemiología y el comportamiento de estas enfermedades18, y se encuentran diferencias con lo publicado en la literatura global.
En Colombia se han publicado algunos reportes de caso19–22 y estudios descriptivos23,24 acerca de estas entidades. En los últimos años ha cambiado su clasificación, concepción y tratamiento, por lo cual resulta relevante y necesario obtener información actualizada de estas enfermedades. El objetivo del presente estudio fue describir las características demográficas y clínicas de los pacientes con MII en 2 instituciones de alta complejidad.
Materiales y métodosDiseño del estudio y selección de los pacientesSe llevó a cabo un estudio observacional, descriptivo, retrospectivo de una cohorte de pacientes con MII admitidos en 2 centros de alta complejidad entre los años 2010 y 2015.
Se incluyeron pacientes mayores de 18 años, con diagnóstico de DM o PM realizado por médico reumatólogo o que cumpliera con los criterios de Bohan y Peter modificados6,25–28. Se excluyeron pacientes que tuvieran diagnóstico de otra enfermedad reumatológica.
Proceso de recolección de la informaciónLa información del estudio se obtuvo mediante la revisión de historias clínicas físicas y electrónicas de los pacientes que cumplieron con los criterios de inclusión, los datos fueron registrados en un formulario electrónico diseñado con la herramienta MAGPI, según las variables necesarias para cumplir con los objetivos del estudio. Se hizo una prueba piloto con 12 historias clínicas, con el fin de estandarizar el proceso de recolección, verificar la calidad de los datos registrados y realizar posibles ajustes al formulario.
En el caso de las variables no encontradas en los registros de historia clínica seleccionados, se llevó a cabo una búsqueda en registros de enfermería, resultados de laboratorio y valoraciones hechas por otras especialidades.
Las variables recolectadas fueron las siguientes:
- •
Demográficas: edad, sexo, fecha de nacimiento y edad en años al momento del diagnóstico.
- •
Antropométricas: peso, talla e índice de masa corporal.
- •
Características clínicas: compromiso muscular (distal o proximal), escala de fuerza muscular (según Medical Research Council29) y comorbilidades (neoplasias, infecciosas, cardiovasculares, sistémicas).
- •
Se clasificaron las MII según los criterios de Bohan y Peter modificados30. Para la miopatía por cuerpos de inclusión, se tuvieron en cuenta los criterios del Centro Europeo Neuromuscular (ENMC, por sus siglas en inglés) 201131, mientras que en el caso de la miopatía necrosante se tuvieron en cuenta hallazgos clínicos y la relación temporal con el uso de medicamentos32.
- •
Desenlaces clínicos: hospitalización, ingreso a unidad de cuidados intensivos, muerte, infecciones recurrentes (definidas como más de un episodio de infección en los últimos seis meses que hubiera requerido manejo hospitalario), complicaciones, limitación funcional determinada por la clase funcional de Steinbroker33, deterioro de la función renal (incremento mayor a 0,3mg/dl/en 48h o más de 1,5 veces el valor de la creatinina basal)34, refractariedad al tratamiento (definida como falla para lograr remisión de la enfermedad luego de una dosis de 0,5mg/kg de metilprednisolona durante un mes, con desmonte progresivo en los siguientes 3 meses o incapacidad para obtener mejoría luego de tratar con terapia inmunosupresora de segunda línea o inmunoglobulina)35.
- •
Laboratorio: creatincinasa total (CPK), lactato deshidrogenasa (LDH), aldolasa, transaminasas (AST, ALT), pruebas de autoinmunidad (ANA, anti-ENA, anti Jo-1).
- •
Ayudas diagnósticas: resonancia magnética nuclear (RMN), electromiografía (EMG) y biopsia muscular.
- •
Tratamiento: glucocorticoides y otros medicamentos inmunomoduladores a inmunosupresores (cloroquina, hidroxicloroquina, metotrexate, micofenolato mofetil, inmunoglobulina intravenosa, ciclosporina, tacrolimus, azatioprina y rituximab) según el tipo de miopatía inflamatoria.
La información fue exportada a una base de datos de Microsoft Excel® 2011, la cual contenía campos de restricción de ingreso de datos para disminuir posibles errores en la digitación; además, se hizo una categorización de las variables cuantitativas según los criterios clínicos. Antes de proceder al análisis de la información, se verificó su consistencia mediante la exploración de valores y la concordancia de los datos registrados. En caso de algún dato confuso, se hizo una nueva revisión de la historia clínica.
Plan de análisisLas variables cualitativas se expresaron mediante frecuencias absolutas y relativas, mientras que para las cuantitativas se emplearon media y desviación estándar (DE), o mediana con sus respectivos rangos intercuartílicos (RIQ) dependiendo de la distribución de los datos. Los análisis estadísticos se llevaron a cabo con el paquete estadístico IBM SPSS® v.22.
Control de sesgosSesgos de selecciónSe controlaron mediante una revisión rigurosa y exhaustiva de las historias clínicas de los pacientes que cumplieran con los criterios de elegibilidad del estudio.
Sesgos de informaciónCon el fin de controlar posibles sesgos de información, se realizó una prueba piloto, se llevaron a cabo reuniones de retroalimentación del proceso de recolección y se diseñó una base de datos utilizando campos con restricciones de ingreso.
Consideraciones éticasSegún la Resolución 8430 de 1993, artículo 11, del Ministerio de Salud de Colombia, esta investigación se considera sin riesgo, ya que se utilizaron métodos documentales retrospectivos realizando una revisión de historias clínicas, con previa aprobación por parte del comité de ética de investigación de las instituciones participantes.
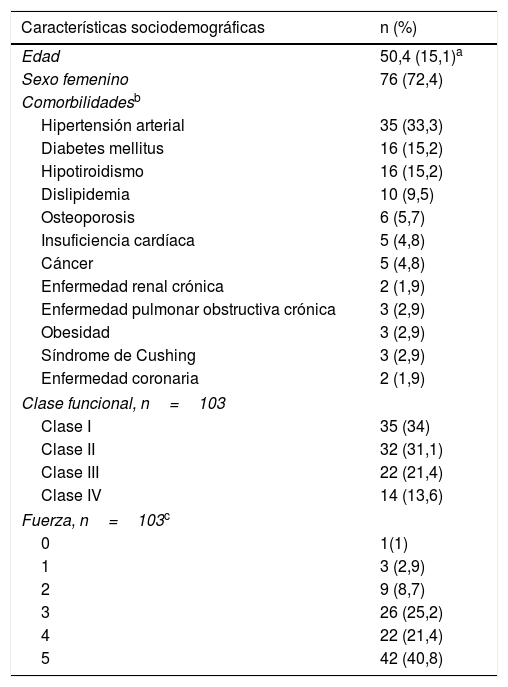
ResultadosCaracterísticas demográficas y comorbilidadesSe revisaron 315 registros clínicos, de los cuales 105 cumplieron criterios de elegibilidad; 76 pacientes (72,4%) fueron mujeres, la edad promedio fue de 50,4 (±15,1) años al momento del ingreso a la cohorte. La comorbilidad más frecuente fue la hipertensión arterial (n=35; 33,3%), seguida por la diabetes mellitus (n=16; 15,2%) y el hipotiroidismo (n=16; 15,2%). En cuanto a la clase funcional al momento de ingresar a la cohorte, la mayoría tenía clases II (31,1%) y III (21,4%). Cincuenta y un sujetos (48,6%) requirieron hospitalización y 7 (6,7%) ingresaron a la unidad de cuidados intensivos (tabla 1).
Características sociodemográficas y clínicas de los pacientes con miopatía inflamatoria
| Características sociodemográficas | n (%) |
|---|---|
| Edad | 50,4 (15,1)a |
| Sexo femenino | 76 (72,4) |
| Comorbilidadesb | |
| Hipertensión arterial | 35 (33,3) |
| Diabetes mellitus | 16 (15,2) |
| Hipotiroidismo | 16 (15,2) |
| Dislipidemia | 10 (9,5) |
| Osteoporosis | 6 (5,7) |
| Insuficiencia cardíaca | 5 (4,8) |
| Cáncer | 5 (4,8) |
| Enfermedad renal crónica | 2 (1,9) |
| Enfermedad pulmonar obstructiva crónica | 3 (2,9) |
| Obesidad | 3 (2,9) |
| Síndrome de Cushing | 3 (2,9) |
| Enfermedad coronaria | 2 (1,9) |
| Clase funcional, n=103 | |
| Clase I | 35 (34) |
| Clase II | 32 (31,1) |
| Clase III | 22 (21,4) |
| Clase IV | 14 (13,6) |
| Fuerza, n=103c | |
| 0 | 1(1) |
| 1 | 3 (2,9) |
| 2 | 9 (8,7) |
| 3 | 26 (25,2) |
| 4 | 22 (21,4) |
| 5 | 42 (40,8) |
La miopatía inflamatoria se clasificó como definitiva en 50 individuos (48,5%). Adicionalmente, 60 pacientes (57,1%) presentaron debilidad muscular simétrica, seguida en frecuencia por trastorno de la deglución (26,7%).
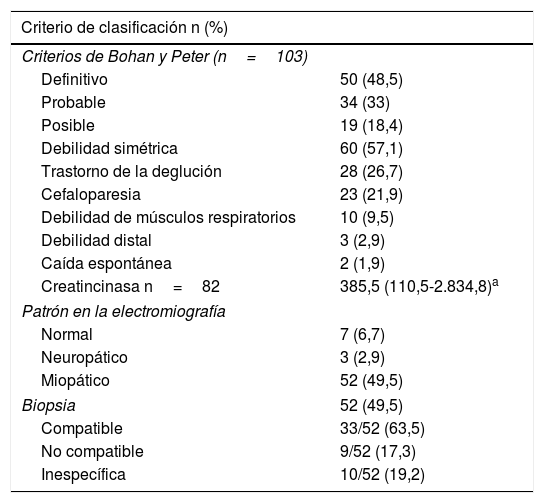
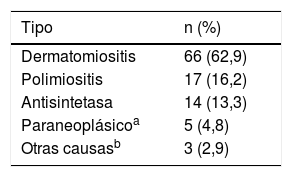
En cuanto a las ayudas diagnósticas, la RMN muscular se llevó a cabo en 44 sujetos (41,9%), de los cuales 8 (18,2%) tuvieron un resultado compatible con miopatía inflamatoria (tabla 2). El tipo de miopatía inflamatoria más frecuente fue la dermatomiositis (n=66; 62,9%) (tabla 3).
Criterios de clasificación, predictores de gravedad y ayudas diagnósticas en una cohorte de miopatía inflamatoria
| Criterio de clasificación n (%) | |
|---|---|
| Criterios de Bohan y Peter (n=103) | |
| Definitivo | 50 (48,5) |
| Probable | 34 (33) |
| Posible | 19 (18,4) |
| Debilidad simétrica | 60 (57,1) |
| Trastorno de la deglución | 28 (26,7) |
| Cefaloparesia | 23 (21,9) |
| Debilidad de músculos respiratorios | 10 (9,5) |
| Debilidad distal | 3 (2,9) |
| Caída espontánea | 2 (1,9) |
| Creatincinasa n=82 | 385,5 (110,5-2.834,8)a |
| Patrón en la electromiografía | |
| Normal | 7 (6,7) |
| Neuropático | 3 (2,9) |
| Miopático | 52 (49,5) |
| Biopsia | 52 (49,5) |
| Compatible | 33/52 (63,5) |
| No compatible | 9/52 (17,3) |
| Inespecífica | 10/52 (19,2) |
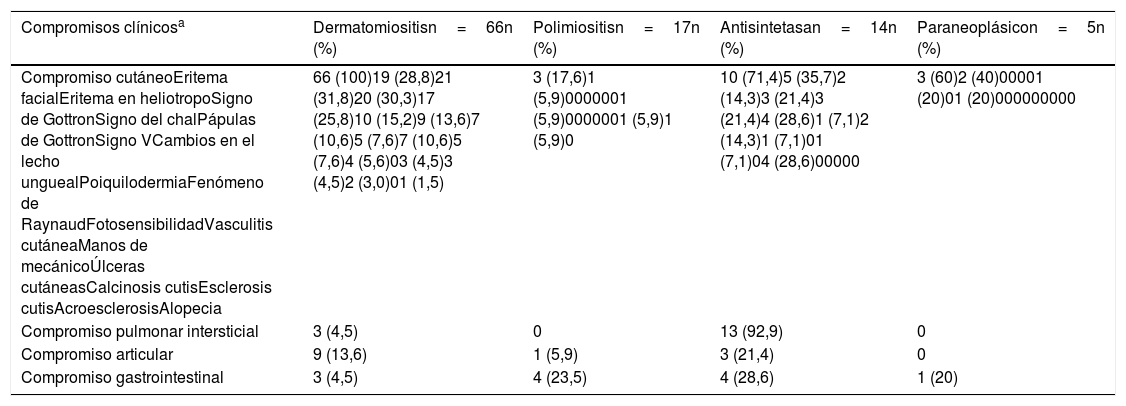
La piel fue el órgano comprometido con mayor frecuencia, especialmente en dermatomiositis (n=66; 100%), y de estas manifestaciones cutáneas, el eritema en heliotropo (n=21; 31,8%) y el signo de Gottron (n=20; 30,3%) fueron los más frecuentes.
En el síndrome antisintetasa el pulmón fue el órgano más afectado (n=13; 92,9%), debido sobre todo a enfermedad pulmonar intersticial, mientras que el compromiso articular se presentó en 3 (21,4%) pacientes (tabla 4).
Características clínicas más frecuentes según tipo de miopatía
| Compromisos clínicosa | Dermatomiositisn=66n (%) | Polimiositisn=17n (%) | Antisintetasan=14n (%) | Paraneoplásicon=5n (%) |
|---|---|---|---|---|
| Compromiso cutáneoEritema facialEritema en heliotropoSigno de GottronSigno del chalPápulas de GottronSigno VCambios en el lecho unguealPoiquilodermiaFenómeno de RaynaudFotosensibilidadVasculitis cutáneaManos de mecánicoÚlceras cutáneasCalcinosis cutisEsclerosis cutisAcroesclerosisAlopecia | 66 (100)19 (28,8)21 (31,8)20 (30,3)17 (25,8)10 (15,2)9 (13,6)7 (10,6)5 (7,6)7 (10,6)5 (7,6)4 (5,6)03 (4,5)3 (4,5)2 (3,0)01 (1,5) | 3 (17,6)1 (5,9)0000001 (5,9)0000001 (5,9)1 (5,9)0 | 10 (71,4)5 (35,7)2 (14,3)3 (21,4)3 (21,4)4 (28,6)1 (7,1)2 (14,3)1 (7,1)01 (7,1)04 (28,6)00000 | 3 (60)2 (40)00001 (20)01 (20)000000000 |
| Compromiso pulmonar intersticial | 3 (4,5) | 0 | 13 (92,9) | 0 |
| Compromiso articular | 9 (13,6) | 1 (5,9) | 3 (21,4) | 0 |
| Compromiso gastrointestinal | 3 (4,5) | 4 (23,5) | 4 (28,6) | 1 (20) |
a Los pacientes pueden tener compromiso de uno o más órganos.
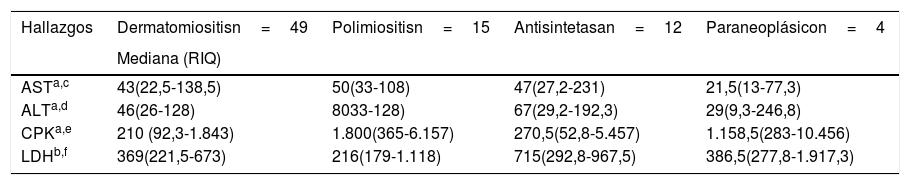
Las enzimas musculares que más presentaron elevación en todos los grupos de miopatías fueron la CPK total y la LDH. La CPK total tuvo mayor elevación en polimiositis, con una mediana de 1.800UI/l (365-6.157), mientras que para la LDH se registró mayor elevación en el síndrome antisintetasa, con una mediana de 715UI/l (292,8-967,5). Los anticuerpos anti-Ro fueron positivos en el 50% de los pacientes con síndrome antisintetasa y en el 11,1% de individuos con polimiositis. En la tabla 5 se presentan otros resultados.
Hallazgos de laboratorio por tipo de miopatía
| Hallazgos | Dermatomiositisn=49 | Polimiositisn=15 | Antisintetasan=12 | Paraneoplásicon=4 |
|---|---|---|---|---|
| Mediana (RIQ) | ||||
| ASTa,c | 43(22,5-138,5) | 50(33-108) | 47(27,2-231) | 21,5(13-77,3) |
| ALTa,d | 46(26-128) | 8033-128) | 67(29,2-192,3) | 29(9,3-246,8) |
| CPKa,e | 210 (92,3-1.843) | 1.800(365-6.157) | 270,5(52,8-5.457) | 1.158,5(283-10.456) |
| LDHb,f | 369(221,5-673) | 216(179-1.118) | 715(292,8-967,5) | 386,5(277,8-1.917,3) |
| n/N (%) | n/N (%) | n/N (%) | n/N (%) | |
|---|---|---|---|---|
| ANA | 38/46 (82,6) | 8/11 (72,7) | 5/9 (55,6) | 0 |
| Patrón ANA | ||||
| Moteado | 23/30 (76,7) | 2/8 (25) | 3/5 (60) | |
| Homogéneo | 4/30 (13,3) | 6/8 (75) | 1/5 (20) | |
| Nucleolar | 1/30 (3,3) | 0 | 1/5 (20) | |
| Citoplasmático | 2/30 (6,7) | 0 | 0 | |
| Anti-Jo1 positivo | 0 | 0 | 4/8 (50) | 0 |
| Anti-Ro positivo | 3/35 (8,6) | 1/9 (11,1) | 5/10 (50) | 0 |
ANA: anticuerpos antinucleares; ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; CPK: creatincinasa total; LDH: lactato deshidrogenasa; RIQ: rangos intercuartílicos.
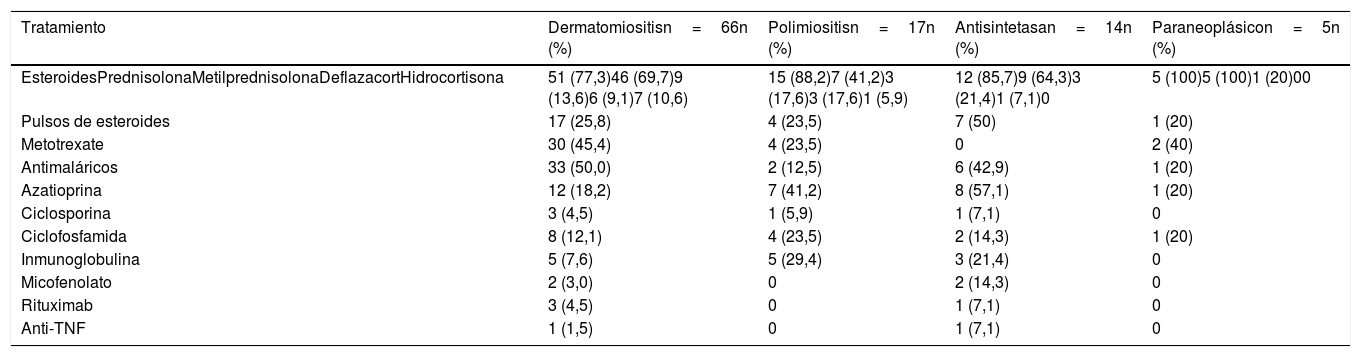
Los esteroides, utilizados en 83 (81,3%) sujetos, fueron el principal tratamiento en todos los tipos de miopatías. El esteroide usado con mayor frecuencia fue la prednisolona (n=67; 65,7%), seguido por la metilprednisolona (n=16; 15,7%). Siete (50%) individuos con síndrome antisintetasa requirieron pulsos de esteroides, mientras que en el grupo de miopatías paraneoplásicas solo uno requirió esta terapia.
Por otra parte, los antimaláricos fueron usados con mayor frecuencia en los pacientes con dermatomiositis (n=33; 50%); la azatioprina (AZA) se utilizó en 8 (57,1%) de los pacientes con síndrome antisintetasa. Finalmente, el micofenolato mofetil (MF) se usó en 2 pacientes con síndrome antisintetasa. El metotrexate (MTX) se empleó en 36 (33,3%) pacientes de todos los tipos de miopatía. El resto del manejo inmunosupresor se describe en la tabla 6. Dos pacientes recibieron anti-TNF por miopatía inflamatoria refractaria: una paciente con dermatomiositis tratada con adalimumab y un paciente con síndrome antisintetasa tratado con etanercept.
Tratamiento según tipo de miopatía
| Tratamiento | Dermatomiositisn=66n (%) | Polimiositisn=17n (%) | Antisintetasan=14n (%) | Paraneoplásicon=5n (%) |
|---|---|---|---|---|
| EsteroidesPrednisolonaMetilprednisolonaDeflazacortHidrocortisona | 51 (77,3)46 (69,7)9 (13,6)6 (9,1)7 (10,6) | 15 (88,2)7 (41,2)3 (17,6)3 (17,6)1 (5,9) | 12 (85,7)9 (64,3)3 (21,4)1 (7,1)0 | 5 (100)5 (100)1 (20)00 |
| Pulsos de esteroides | 17 (25,8) | 4 (23,5) | 7 (50) | 1 (20) |
| Metotrexate | 30 (45,4) | 4 (23,5) | 0 | 2 (40) |
| Antimaláricos | 33 (50,0) | 2 (12,5) | 6 (42,9) | 1 (20) |
| Azatioprina | 12 (18,2) | 7 (41,2) | 8 (57,1) | 1 (20) |
| Ciclosporina | 3 (4,5) | 1 (5,9) | 1 (7,1) | 0 |
| Ciclofosfamida | 8 (12,1) | 4 (23,5) | 2 (14,3) | 1 (20) |
| Inmunoglobulina | 5 (7,6) | 5 (29,4) | 3 (21,4) | 0 |
| Micofenolato | 2 (3,0) | 0 | 2 (14,3) | 0 |
| Rituximab | 3 (4,5) | 0 | 1 (7,1) | 0 |
| Anti-TNF | 1 (1,5) | 0 | 1 (7,1) | 0 |
TNF: factor de necrosis tumoral.
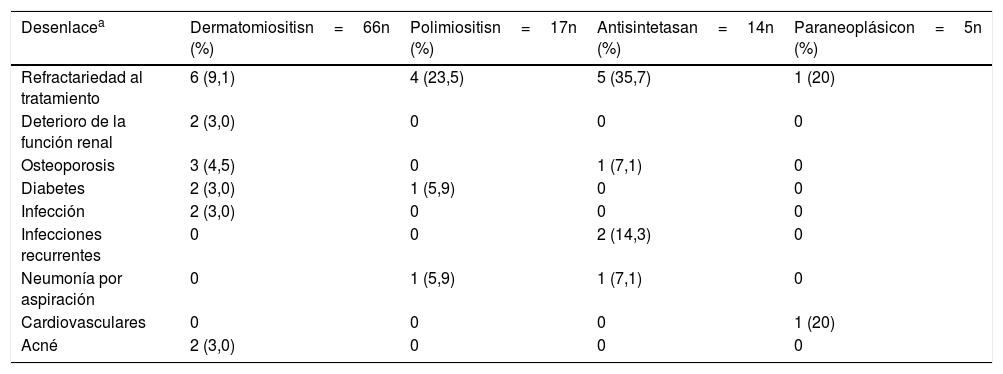
La complicación más frecuente fue la refractariedad al tratamiento inmunosupresor, principalmente en síndrome antisintetasa (n=5; 35,7%) y en la polimiositis (n=4; 23,5%); la segunda en frecuencia correspondió a infecciones recurrentes en el grupo de síndrome antisintetasa (n=2; 14,3%), seguida por neumonía por aspiración en síndrome antisintetasa (n=1; 7,1%) y en polimiositis (n=1; 5,9%) (tabla 7). Dos pacientes presentaron compromiso renal, el cual fue explicado por otras etiologías diferentes a miopatías inflamatorias.
Desenlaces clínicos según tipo de miopatía
| Desenlacea | Dermatomiositisn=66n (%) | Polimiositisn=17n (%) | Antisintetasan=14n (%) | Paraneoplásicon=5n (%) |
|---|---|---|---|---|
| Refractariedad al tratamiento | 6 (9,1) | 4 (23,5) | 5 (35,7) | 1 (20) |
| Deterioro de la función renal | 2 (3,0) | 0 | 0 | 0 |
| Osteoporosis | 3 (4,5) | 0 | 1 (7,1) | 0 |
| Diabetes | 2 (3,0) | 1 (5,9) | 0 | 0 |
| Infección | 2 (3,0) | 0 | 0 | 0 |
| Infecciones recurrentes | 0 | 0 | 2 (14,3) | 0 |
| Neumonía por aspiración | 0 | 1 (5,9) | 1 (7,1) | 0 |
| Cardiovasculares | 0 | 0 | 0 | 1 (20) |
| Acné | 2 (3,0) | 0 | 0 | 0 |
Cinco pacientes (4,8%) fallecieron durante la primera valoración, de los cuales 2 tenían miopatía paraneoplásica, uno dermatomiositis, uno polimiositis y otro síndrome antisintetasa; 4 de estos pacientes fallecieron por complicaciones infecciosas como neumonía y bacteriemia, mientras que un paciente falleció por paro cardiorrespiratorio secundario a falla respiratoria.
DiscusiónHasta donde se tiene conocimiento, este es el estudio más amplio sobre miopatías inflamatorias en Colombia.
Se encontró una mayor proporción de mujeres afectadas, lo cual concuerda con otros estudios locales e internacionales12,13,15,23,36.
Dentro de las comorbilidades, los hallazgos son similares a otras series de pacientes37–39.
Por otro lado, se destaca la importante cantidad de pacientes que requirieron ingreso a hospitalización y unidad de cuidados intensivos, lo que es indicio de un curso crónico, progresivo y grave. Otros autores también han descrito una mayor tasa de hospitalización en pacientes con miopatías inflamatorias40,41, lo cual se relaciona con el curso progresivo de la enfermedad40.
En la actualidad existen varios criterios de clasificación para las miopatías inflamatorias (Bohan y Peter27,28, Tanimoto et al.42, Targoff et al.43, Dalakas y Hohlfeld2, ENMC30), sin embargo, la mayoría no han sido ampliamente validados. Los más utilizados en los diferentes estudios clínicos son los de Bohan y Peter, con una alta sensibilidad (98%) y baja especificidad (55%)27,28. En nuestro estudio, el diagnóstico de miopatía inflamatoria fue realizado por criterio clínico del reumatólogo tratante, en tanto que los pacientes fueron clasificados de acuerdo con los criterios de Bohan y Peter modificados. Recientemente, se publicaron los nuevos criterios clasificatorios American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR), los cuales han demostrado tener una buena correlación con los criterios de Bohan y Peter ampliamente utilizados, con una sensibilidad del 93% y una especificidad del 88% cuando se tiene información de biopsia44. Lo anterior muestra que los criterios de clasificación utilizados en la cohorte tienen un desempeño adecuado, incluso al compararlos con los nuevos criterios.
El compromiso muscular en la mayoría de los pacientes se dio por debilidad proximal y simétrica. En el estudio realizado por Pinto et al.23 se encontró debilidad muscular en cintura pélvica hasta en el 80% de los pacientes, y se hicieron hallazgos cutáneos en dermatomiositis similares a los descritos en nuestra cohorte, excepto por una mayor frecuencia de fenómeno de Raynaud, cambios vasculíticos, manos de mecánico, poiquilodermia y calcinosis cutis.
Otras manifestaciones extramusculares como la disfagia, el compromiso articular y la enfermedad pulmonar intersticial difusa (EPID) fueron similares a los hallazgos hechos por Pinto et al.23. Por otro lado, en un estudio publicado por Dobloug et al. se encontró debilidad muscular como hallazgo predominante en el 84% de los pacientes, disnea en el 44% de DM y el 22% de PM, disfagia en el 23% y fenómeno de Raynaud junto con manos de mecánico, que fueron más frecuentes en DM45. Al comparar lo descrito anteriormente con los datos reportados por el registro EuroMyositis13, se puede apreciar que hubo similitud en la debilidad muscular como síntoma cardinal de los diferentes subtipos de miopatías; sin embargo, en dicho registro hubo mayor frecuencia de algunas manifestaciones cutáneas como fenómeno de Raynaud, manos de mecánico, calcinosis cutis y úlceras cutáneas; mientras que en el SAS hubo mayor compromiso articular que en lo descrito en nuestra cohorte.
Con respecto a las ayudas de laboratorio, debe aclararse que solo se encuentran algunos anticuerpos específicos de miositis (anti-Jo1, y anti-Pm/Scl); no obstante, en la descripción de los criterios clasificatorios ACR/EULAR30 también se puso en evidencia la falta de disponibilidad de los datos de anticuerpos específicos.
Al evaluar los resultados de laboratorio resulta llamativo que la mitad de los pacientes con antisintetasa tuvieron positividad de anti-Ro y anti-Jo1. Koenig et al.46 mostraron que los anticuerpos anti-Jo1 se encontraron de manera concomitante con los anti-Ro en un 73%, en tanto que se halló una menor respuesta a la prednisolona y un mayor requerimiento de terapia de segunda línea entre aquellos pacientes con positividad de anti-Jo1, lo cual podría ser una explicación de por qué en nuestra cohorte el subgrupo de antisintetasa fue más refractario al tratamiento y requirió mayor cantidad de pulsos de esteroides.
En nuestra serie, las enzimas musculares que más presentaron elevación fueron la CPK total y la LDH; sin embargo, existen algunas diferencias con respecto a otras publicaciones de Noruega45 y España37 en las que se encontraron mayores niveles de CPK total en pacientes con dermatomiositis, polimiositis y síndrome paraneoplásico. Algunas hipótesis podrían explicar tal hallazgo, como el hecho de que muchos pacientes venían con diagnóstico y tratamiento inmunosupresor previamente al ingreso al estudio, y es posible que esto disminuya los niveles de CPK total. Otra explicación de estos hallazgos es que los pacientes con MII de larga duración pueden presentarse con valores de CPK total cercanos a lo normal, lo cual ocurre cuando la mayor parte del músculo ha sido afectada y se reemplaza por tejido graso. También debe reconocerse la baja especificidad de la LDH en miopatías inflamatorias47.
Lo descrito en el presente estudio es concordante con los esquemas de tratamiento analizados por Meyer et al.48. Otros estudios, en su mayoría cohortes retrospectivas13,37,49, series de casos50 y algunos ensayos clínicos51–54, han descrito el uso de esteroides como terapia de primera línea aunado a otros inmunosupresores8,50. En una publicación realizada por Nuño et al.37, los esquemas de tratamiento utilizados fueron similares a los encontrados en el presente trabajo. Llamó la atención el uso de anti-TNF en 2 pacientes de la cohorte (uno con dermatomiositis y otro con síndrome antisintetasa), en los cuales estos medicamentos se utilizaron por enfermedad refractaria. En ninguno de los 2 casos hubo respuesta; además, existe evidencia de recaídas frecuentes55 con su uso y la posible inducción y el empeoramiento de la miopatía inflamatoria y el compromiso pulmonar en estas entidades56.
Los hallazgos relacionados con complicaciones y mortalidad fueron semejantes a complicaciones infecciosas descritas en un estudio publicado por Murray et al.57, en el cual se reportó que, de las 15.407 hospitalizaciones por DM y PM, el 4,5% de los pacientes murió y las principales causas de mortalidad fueron neumonía e infección. Las infecciones en pacientes con MII pueden ser explicadas por el mayor riesgo de infecciones bacterianas y fúngicas, incluso en ausencia de manejo inmunosupresor, lo cual sugiere defectos en la inmunidad mediada por células que predisponen a infecciones. Asimismo, se presentaron menos muertes con respecto a otros estudios23,49,58, dado que se trata de la descripción del ingreso de los pacientes a la cohorte y no hay datos de seguimiento en el tiempo.
Este estudio tiene varias limitaciones: fue inevitable el sesgo de selección, debido a que los centros participantes tienen un alto nivel de complejidad y es posible que los pacientes tuvieran mayor gravedad de la enfermedad o fueran ampliamente estudiados. Adicionalmente, puede haber sesgos de información asociados al diseño retrospectivo del estudio, pero se trató de controlarlos con las estrategias descritas en materiales y métodos.
Como fortalezas de este trabajo se destacan el importante número de pacientes incluidos y los criterios clasificatorios actualizados.
ConclusionesEn una cohorte de pacientes con miopatías inflamatorias del noroccidente colombiano se encontró una frecuencia importante de hospitalización y de ingreso a la unidad de cuidados intensivos, lo cual tiene relación con mayor gravedad de la enfermedad y refractariedad al tratamiento. Asimismo, hubo un número importante de pacientes con síndrome antisintetasa, quienes a su vez presentaron mayor refractariedad al tratamiento.
Conflicto de interésLos autores declaran no tener ningún conflicto de intereses.
A las instituciones participantes.
