



La crisis renal esclerodérmica (CRS) es una manifestación rara de la esclerosis sistémica (ES). Se presenta como hipertensión arterial de nuevo inicio, empeoramiento o aceleración de la hipertensión arterial crónica o rápido deterioro de la función renal, frecuentemente acompañada de signos de hemólisis microangiopática. Su relación con el síndrome hemolítico urémico es infrecuente, existiendo tan solo un caso similar reportado en la literatura. Presentamos el caso de una mujer de 36 años en tratamiento para ES con cefalea global, pulsátil e intensa, convulsión tónico-clónica, cifras de presión arterial altas, falla renal aguda y hemólisis no autoinmune persistente. La evaluación de ADAMTS13 mostró un 60,6% de actividad. El estudio genético para búsqueda de mutaciones predisponentes para síndrome hemolítico urémico atípico (SHUa) reveló variante homocigota en el gen ADAMTS13, c.3287G>A (p.Arg1096His). Se inició tratamiento con eculizumab, observándose en poco tiempo mejoría de la hemólisis, función renal y estado clínico, con algunos efectos benéficos notorios e inesperados sobre la ES.
Scleroderma renal crisis (SRC) is a rare manifestation of systemic sclerosis (SSc), presented as hypertension of new onset, worsening and / or acceleration of chronic hypertension, or rapid deterioration of renal function, often accompanied by signs of microangiopathic haemolysis. It is rarely associated with haemolytic uraemic syndrome, and there is only one similar case reported in the literature. The case is presented here of a 36-year-old woman on treatment for SSc with global, pulsatile and intense headache, clonic tonic convulsions, high blood pressure levels, acute renal failure, and persistent non-autoimmune haemolysis. The evaluation of ADAMTS13 showed 60.6% of activity. The genetic study to search for mutations predisposing to atypical haemolytic uraemic syndrome (aHUS) revealed a homozygous variant in ADAMTS13 gene, c.3287G>A (p.Arg1096His). Eculizumab was started, with an improvement being observed in a short time in the haemolysis, renal function, and clinical status, with some notable and unexpected beneficial effects on SSc.
La esclerosis sistémica (ES) es una enfermedad del tejido conectivo que involucra múltiples órganos, caracterizada por un excesivo depósito de colágeno, autoinmunidad, hiperreactividad vascular y fenómenos microvasculares obliterantes. El daño vascular puede manifestarse como fenómeno de Raynaud, isquemia digital, hipertensión arterial pulmonar o crisis renal esclerodérmica (CRS). Su relación con el síndrome hemolítico urémico es infrecuente, existiendo tan solo un caso similar reportado en la literatura1,2. La CRS es una manifestación rara de la ES que se presenta como hipertensión arterial de nuevo inicio, empeoramiento o aceleración de la hipertensión arterial crónica o rápido deterioro de la función renal, frecuentemente acompañada de signos de hemólisis microangiopática3. Actualmente la CRS se desarrolla en el 5-10% de los pacientes, principalmente en la ES difusa4.
Presentamos un caso de falla renal aguda con microangiopatía trombótica (MAT), anemia hemolítica microangiopática, en una paciente con sobreposición de lupus eritematoso sistémico y ES. Lo interesante de este caso es la resolución de la hemólisis microangiopática con el uso de anticuerpos monoclonales anti-C5 (eculizumab).
Presentación del casoUna mujer blanca de 36 años ingresó al servicio de urgencias con 14 horas de cefalea global, pulsátil e intensa, seguida de convulsión tónico-clónica generalizada y presión arterial de 195/120mmHg. Su evaluación inicial reveló frecuencia cardiaca (FC) de 100 latidos/minuto, frecuencia respiratoria (FR) de 25 respiraciones/minuto, temperatura de 36,2̊C, oximetría de pulso 91% con FiO2 0,21, desorientación temporoespacial sin déficit neurológico focal, sin signos meníngeos y amnesia del evento.
Al examen físico presentaba microstomía y esclerodactilia sin otros hallazgos relevantes. Había sido diagnosticada en diciembre de 2015 con síndrome de sobreposición con posible lupus eritematoso sistémico, sustentado en episodios de serositis recurrente (discreto derrame pericárdico y pleural), leucopenia, anticuerpos antinucleares (ANA) a título 1:640 de patrón homogéneo y consumo del complemento más un componente escleriforme documentado por esclerodactilia, neumonía intersticial no específica (NINE), microstomía y anti-Scl 70 positivo a título 108,6 (ES forma difusa). Recibía tratamiento con cloroquina 250mg/día y prednisona 1mg/kg/día (60mg/día), metotrexato 15mg/semana como ahorrador de esteroide y colchicina.
Los análisis de laboratorio a la admisión incluyeron hemograma con 15.520 leucocitos, 13.270 neutrófilos, 1.580 linfocitos, hemoglobina 9,5g/dl, hematocrito 30%, volumen corpuscular medio 97fl, ancho de distribución eritrocitaria 18,3%, 100.000 plaquetas, creatinina 6,7mg/dl, nitrógeno ureico 78,9mg/dl, potasio 4,3mEq/l, sodio 132mEq/l, cloro 102mmol/l, calcio 8,9mg/dl, bilirrubina total 1,0mg/dl, bilirrubina directa e indirecta normales, TGO 33U/l, TGP 15U/l, fosfatasa alcalina 57,4UI/l, glucemia 89mg/dl, proteína C reactiva 125,8mg/l, frotis de sangre periférica con fórmula leucocitaria normal, morfología globular blanca normal, 89% neutrófilos, 5% linfocitos, 6% monocitos, morfología eritrocitaria con anisocitosis: microcitos+, policromatofilia: poiquilocitos+, esquistocitos++, plaquetas disminuidas en número con recuento manual 99.000/mm3, reticulocitos 6,5%, deshidrogenasa láctica (LDH) 663U/l, Coombs directo negativo, uroanálisis normal, sin proteinuria, sin sedimento activo, gases arteriales normales, tomografía simple de cráneo con hipodensidad corticosubcortical, algunas lacunares occipitales bilaterales, ecocardiograma transtorácico con reporte de derrame pericárdico de 350ml, válvulas intactas y normofuncionantes, contractilidad global deprimida con fracción de eyección del 30%. Fue diagnosticada con encefalopatía posterior reversible, crisis hipertensiva tipo emergencia, CRS y MAT tipo púrpura trombocitopénica trombótica (PTT) vs. síndrome hemolítico urémico atípico (SHUa). Recibió manejo con labetalol intravenoso, la dosis de prednisona fue disminuida a 15mg/día, nifedipino oral y fueron indicadas 5 terapias de recambio plasmático a 1,5l/día. Su evolución clínica no fue favorable. Aunque revirtió la encefalopatía con mejoría neurológica completa y se logró retirar la infusión de labetalol manteniendo el control de la presión arterial, el compromiso cutáneo progresó rápidamente avanzando hasta escleroderma franca con Rodnan score de 22 y fenómeno de Raynaud, capilaroscopia con longitud anormal, diámetro anormal, tortuosidades, angiogénesis+, megacapilares+, hallazgos compatibles con fase escleriforme tardía. Además, lesión renal con progreso hasta la anuria requiriendo el inicio de terapia de soporte renal con hemodiálisis. El manejo antifibrótico pulmonar fue ajustado a micofenolato mofetilo 3g/día; adicionalmente continuaba presentando anemización, elevación de LDH, haptoglobina baja, esquistocitos en sangre periférica, anti-RNP, anti-SM, anti-RO, anti-LA, C3, C4, P-ANCA, C-ANCA, VHB, VHC, anticardiolipina IgG, IgM, antifosfolípido IgG, IgM, antinucleosoma fueron negativos.
ADAMTS13: 60,6% de actividad. El estudio genético para búsqueda de mutaciones predisponentes para SHUa reveló variante homocigota en el gen ADAMTS13, c.3287G>A (p.Arg1096His) encontrado en un nucleótido conservado y posición de aminoácidos débilmente conservadas con pequeñas diferencias fisicoquímicas entre los aminoácidos intercambiados (Alamut v.2.7.1), C3, CD46, CFB, CFH, CFHR1, CFHR2, CFHR3, CFHR5, CFI, DGKE, PIGA, THBD no tuvieron variaciones patológicas, CFH, CFHR1, CFHR2, CFHR3, CFHR5 no tuvieron deleciones o duplicaciones.
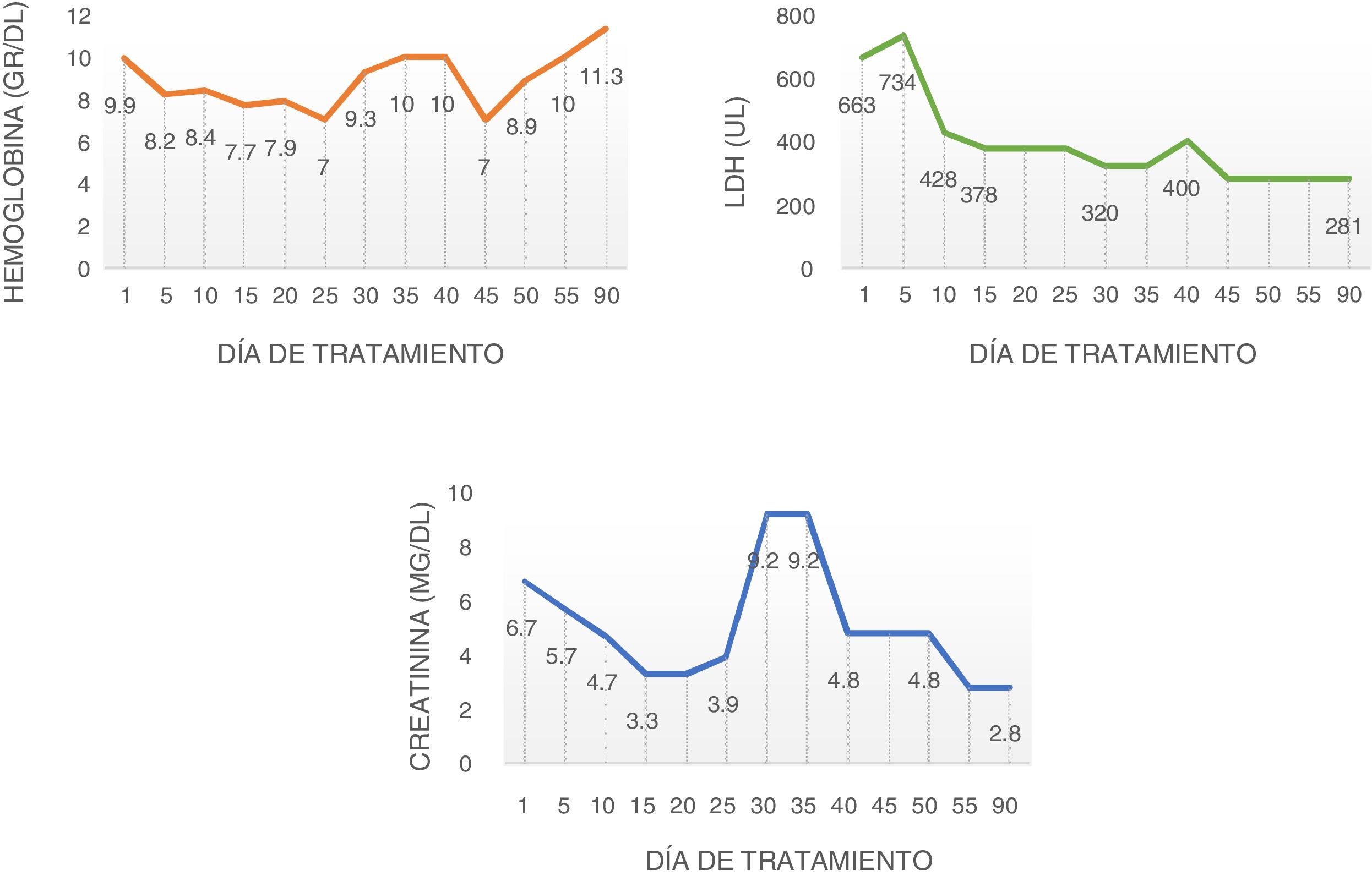
La ecografía renal mostró aumento de la ecogenicidad de la corteza renal. Se trató de demostrar el fenómeno trombótico a nivel renal, pero la biopsia renal no fue concluyente debido a una muestra insuficiente. Ante el deterioro persistente se decidió la vacunación antimeningocócica, antineumocócica y la administración de eculizumab. Tras la dosis de inducción se estabilizaron los niveles de hemoglobina, se detuvo la hemólisis y disminuyó la LDH logrando ser dada de alta 2 semanas después de la inducción (fig. 1). Presentó efecto adverso paradójico constitucional con la administración de micofenolato, lo que obligó a su retiro, por lo cual no recibió otro antifibrótico. En el seguimiento a 3 meses se encontró con normalización de las cifras de presión arterial y los antihipertensivos fueron retirados, recibía tratamiento solo con eculizumab siendo llamativa la estabilización e incluso reversión del compromiso cutáneo, con nueva evaluación del Rodnan score: 14; también presentó mejoría del fenómeno de Raynaud, además de recuperación cardiovascular con ecocardiograma transtorácico de control que revelaba mejoría en la contractilidad global, resolución del derrame pericárdico, fracción de eyección hasta el 64% y mejoría de la disnea; tomografía de tórax de alta resolución de control sin variaciones respecto a la previa.
Discusión
La ES se presenta con una relación mujer:hombre de 3-8:1 y un pico de incidencia entre los 45 y 64 años. Al parecer hay una mayor frecuencia de la enfermedad en mujeres de raza negra. Los criterios para considerar una CRS incluyen hipertensión grave, insuficiencia renal aguda rápidamente progresiva, hemólisis microangiopática y trombocitopenia (<100.000 plaquetas). En el 2016 se actualizó el consenso para los criterios diagnósticos donde la hipertensión arterial fue definida como presión arterial >150/85mmHg, elevación de la presión arterial sistólica >20mmHg respecto al valor basal del paciente en 2 mediciones durante 24 horas. Las manifestaciones asociadas se definieron como elevación de la creatinina 50% sobre el nivel basal o un aumento absoluto de 0,3mg/dl (26,5μmol/l), proteinuria >2+, hematuria >1+ por tirilla o >10 células rojas por campo de alto poder5. La hipertensión arterial se presenta hasta en el 90% de los pacientes; los hallazgos clínicos más frecuentes son la cefalea, visión borrosa y signos de encefalopatía.
Otras manifestaciones incluyen: ataque cerebrovascular isquémico, retinopatía hipertensiva, falla cardiaca, edema pulmonar, pericarditis, arritmias cardiacas, oliguria y malestar general. Los sangrados intracerebrales son raros. El uso rutinario de inhibidores de la enzima convertidora de angiotensina (ACEIs) ha mejorado notoriamente los resultados en los últimos años; sin embargo, la CRS permanece como una manifestación devastadora de la enfermedad y los resultados funcionales así como la sobrevida son pobres, alcanzando en las mejores series de casos el 70,9% a un año, el 66,6% a 2 años, el 60% a 5 años y el 41,9% a 10 años6,7. Se han descrito algunos factores de riesgo para el desarrollo de CRS, entre ellos: anemia, miocarditis, falla cardiaca, arritmias cardiacas, anti-SCL70 positivo, uso de esteroides (prednisona más de 15mg/día o su equivalente) hasta 6 meses antes del inicio del cuadro; así mismo, se han reportado casos con el uso de ciclosporina. Los hallazgos de laboratorio más frecuentes son: elevación de la creatinina que puede persistir por varios días, incluso tras lograr el control de la hipertensión arterial, proteinuria en rangos de 0,5-2,5g/l, hematuria microscópica, hemoglobinuria, anemia y trombocitopenia que configuran MAT en el 43% de los pacientes, la trombocitopenia generalmente es leve a moderada, rara vez por debajo de 50.000 plaquetas/mm3 y característicamente la MAT resuelve al controlar la hipertensión arterial. Los anti-SCL70 son encontrados hasta en el 33% de los casos, anticuerpos anticentrómero en el 1-3% de los casos. Aún es controversial la ventaja de obtener una biopsia renal; ciertamente no es esencial en el diagnóstico, pero puede ofrecer algunas ventajas, entre ellas excluir el diagnóstico diferencial y evaluar el pronóstico. Cuando la MAT persiste debe realizarse un complejo análisis para tratar de diferenciar la MAT de la CRS de otras causas de MAT, entre ellas: PTT y SHUa. La MAT mediada por disregulación del complemento implica mecanismos genéticos y adquiridos; la cascada del complemento es el pilar de la inmunidad innata e implica un delicado balance entre la actividad y el control constante del complemento. Cuando este balance se rompe sobreviene el daño del SHUa y su patogénesis obedece a una disregulación de la vía alternativa del complemento (VAC). Los reguladores de este proceso incluyen el factor H (FH), el factor I (FI) y el cofactor proteína de membrana (MCP). Dentro de los mecanismos genéticos del SHUa múltiples estudios han identificado mutaciones en genes del complemento hasta en el 60% de los pacientes con SHUa; las mutaciones frecuentemente se presentan con heterocigocidad y penetración del fenotipo hasta del 50%. La más frecuente de las mutaciones en el SHUa es la del FH, que ha sido encontrada en el 25% de los casos esporádicos y en el 40% de los casos familiares.
Presentamos un caso difícil y complejo en una mujer colombiana, blanca, adulta joven, que tras casi 2 años de estudio había sido diagnosticada con síndrome de sobreposición que progresivamente fue variando hacia ES forma difusa y desarrolló episodio de emergencia hipertensiva que era compatible con CRS puesto que se encontraron elementos clínicos, hallazgos de laboratorio y antecedentes exposicionales, entre ellos: el curso dentro del primer año de evolución, elevación súbita de la presión arterial, hallazgos de laboratorio altamente sugestivos de MAT y uso de dosis altas de esteroides en quien se descartaron otras causas de fallo renal.
Si bien los criterios del consenso de CRS de 2016 para definir hipertensión arterial y falla renal son arbitrarios y no se correlacionan con las actuales guías para hipertensión arterial crónica y falla renal, nuestra paciente no presentaba antecedente de hipertensión arterial sistémica, lo que hacía más factible el diagnóstico. Tras recibir asistencia, los niveles de presión arterial fueron controlados y la sintomatología neurológica así como el deterioro cardiopulmonar resolvieron. Sin embargo, persistió la MAT, lo que nos llevó a enfrentar la necesidad de establecer en el campo clínico real cuánto puede perdurar este hallazgo en el contexto de CRS. Desafortunadamente no existe claridad al respecto y no logramos encontrar literatura médica que responda a la pregunta de forma precisa; aun así la gran mayoría de reportes señalan que al controlar la presión arterial y limitarla a valores normales el fenómeno microangiopático debe desaparecer. Con la respuesta clínica observada, la progresiva anemia, trombocitopenia, elevación de LDH y persistencia de esquistocitos buscamos de manera activa diagnósticos diferenciales explorando la posibilidad de PTT o SHUa, situación prioritaria para definir la posibilidad de supervivencia de la paciente. A pesar de que la gran mayoría de reportes sugieren que en el caso de PTT las trombocitopenias observadas suelen ser profundas y los conteos de plaquetas menores de 50.000, los niveles de plaquetas no son un indicador real que pueda ayudar a diferenciar PTT de SHUa, y este único parámetro no debería causar la toma de decisiones terapéuticas; por el contrario, el nivel de actividad de ADAMTS13 sí es un excelente marcador de condiciones innatas y adquiridas que pueden ayudar a diferenciar PTT. En nuestro caso la actividad de ADAMTS13 fue claramente mayor del 5% (60,6%), lo que sugería que se tratara de SHUa. El estudio genético reveló una variante homocigota en el gen ADAMTS13; como hemos mencionado previamente se sabe que varios genes están asociados a SHUa. Esta predisposición se hereda de forma autosómica recesiva o autosómica dominante con penetrancia incompleta; rara vez la herencia es digénica y la aparición del SHUa varía desde la niñez hasta la adultez8. Las variantes del gen de ADAMTS13 son causantes de PTT familiar; sin embargo, han sido también descritas en pacientes con SHUa por Feng et al.9, en especial en combinación con otra variante (A747V). Los análisis funcionales de estas revelan diferencia significativa en comparación con ADAMTS13 recombinante normal. Esta variante es considerada un hallazgo común en pacientes con SHUa y se presenta con una frecuencia de 0,01288, es decir, en 1.546 entre 120.050 alelos y puede elevarse hasta 0,1128 para población latina o disminuir hasta 0,0034 en población europea-americana y 0,0023 en población africana10. Basados en estos hallazgos, en nuestra paciente no puede ser confirmada una predisposición genética al SHUa, pero sí es posible una susceptibilidad incrementada, lo cual se ha correlacionado con la mejoría clínica tras la administración de terapia anticomplemento con eculizumab (anticuerpo monoclonal dirigido contra C5).
ConclusiónLa presentación de nuestro caso busca demostrar la enorme complejidad del diagnóstico de la CRS, patología ya por sí misma rara y que puede participar en sujetos con susceptibilidad incrementada como condición amplificadora del complemento desarrollando SHUa que la llevan a un nuevo nivel de enfoque médico en el cual participan el avance y estado del arte en conocimiento, reconocimiento de patrones clínicos, la experticia, el análisis genético y el uso de nuevos fármacos biológicos. Muchas preguntas persisten sin respuesta hasta el momento respecto al uso de terapia anticomplemento, entre ellas: qué tan larga debe ser la terapia, riesgo de infección meningocócica, riesgo de respuesta inmune al biológico, limitaciones en la dosis y frecuencia de la terapia, criterios de mejoría y en nuestro caso especial el hallazgo incidental de una mejoría clínica notoria de las manifestaciones cutáneas y cardiovasculares de la ES forma difusa, sin ninguna terapia adicional al manejo anticomplemento, planteando la pregunta adicional, a futuras investigaciones, de si es posible que se encuentre en la vía del complemento el manejo de la ES.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés.
