



Erdheim Chester disease (ECD) is a rare non-Langerhans cell histiocytosis. It is characterized by the infiltration of various organs and tissues by foamy histiocytes with a heterogeneous clinical course that varies from mild forms to disseminated forms with progressive and lethal behaviour. The case of a patient who presented with a cerebellar syndrome associated with autoimmune pathology is presented. In the course of the disease, refractoriness to glucocorticoids and clinical manifestation with bone involvement in the form of symmetrical osteosclerosis of long bones were determining factors for suspicion of this entity. We reviewed scientific articles through the PubMed metasearch engine with the keywords “erdheim chester disease”, “erdheim chester and nervous system”, and “autoimmunity and erdheim chester disease”, selecting those with greater emphasis on clinical presentation with neurological involvement and associated autoimmune pathology. Advances in the pathogenesis of ECD have allowed us to understand the nature of the disease, as well as the use of targeted therapies. It is interesting to keep this entity in mind, as well as the pathologies with which it is frequently associated, with the objective of an early diagnosis and a better clinical approach.
La enfermedad de Erdheim-Chester (EEC) es una histiocitosis de células no Langerhans infrecuente. Se caracteriza por la infiltración de varios órganos y tejidos por histiocitos espumosos con un curso clínico heterogéneo que varía desde formas leves hasta aquellas diseminadas con un comportamiento progresivo y letal. Se presenta el caso de una paciente que debutó con un síndrome cerebeloso asociado con patología autoinmune. En el curso evolutivo, la refractariedad a glucocorticoides y la manifestación clínica con afectación ósea en forma de osteoesclerosis simétrica de huesos largos fueron determinantes para la sospecha de esta entidad. Revisamos artículos científicos con el metabuscador PubMed, empleando las palabras clave «erdheim chester disease», «erdheim chester and nervous system» y «autoimmunity and erdheim chester disease», y seleccionamos trabajos con un mayor énfasis en la presentación clínica con afectación neurológica y patología autoinmune asociada. Los avances en la patogénesis de la EEC han permitido conocer la naturaleza de la enfermedad, así como la utilización de terapias dirigidas. Interesa tener presente esta entidad, así como las patologías a las que se asocia con frecuencia, con el objetivo de obtener un diagnóstico precoz y un mejor abordaje clínico.
Histiocytosis is a rare disorder characterized by the accumulation of macrophages, dendritic cells, or monocyte-derived cells in various tissues and organs in both adults and children. The current classification divides these disorders into five groups based on clinical, radiological, pathological, genetic, or molecular features.
Erdheim-Chester disease (ECD) belongs to the “L” group (Langerhans), together with Langerhans cell histiocytosis (LCH) and extracutaneous juvenile xanthogranuloma. It is differentiated from these entities by the clinical presentation supported by histology and fundamentally by the immunohistochemical and molecular analysis characteristics of histiocytes. Thus, it is considered a rare clonal hematopoietic disease defined by multiorgan infiltration of foamy histiocytes, chronic inflammation, and fibrosis.
A case of a patient with a neurological condition associated with an autoimmune disorder is presented. After reviewing the available scientific literature although the pathogenesis of this entity is not completely known, the discovery of clonal mutations that activate the mitogen-activated protein kinase (MAPK) pathways has not only allowed to clarify the nature of this disease, but also has therapeutic implications. On the other hand, a high prevalence of associated clinical or biological autoimmunity is evident, which, although it may entail complexity in the diagnosis, represents a new approach to clarify the etiopathogenic mechanisms of this disorder.
Clinical observationA 55-year-old female with a history of probable undifferentiated connective tissue disease based on clinical symptoms of xerophthalmia, oral ulcers, Raynaud's phenomenon, and positive antinuclear antibodies (ANA) 1/640. She presents with gait instability and dysphagia for one year; physical examination depicts moderate dysarthria and hyperreflexia, with appendicular and gait ataxia.
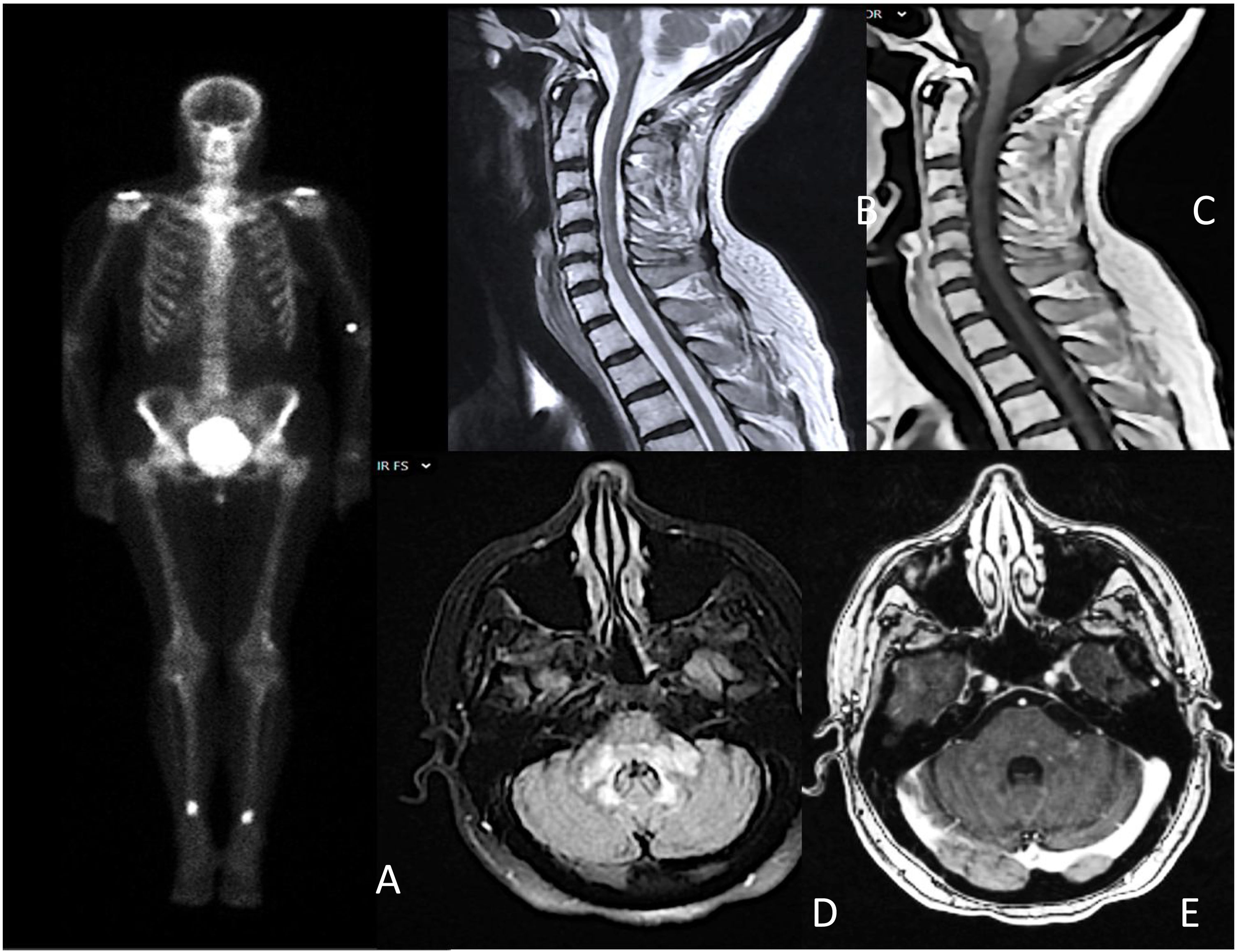
A Schirmer test and a minor salivary gland biopsy fulfilled the European League Against Rheumatism (EULAR) criteria for Sjögren syndrome (SS). Brain MRI revealed hyperintense areas with slight contrast enhancement in the brain stem and extensive longitudinal myelitis at the cervical level (Fig. 1). The remaining studies, including cerebrospinal fluid analysis and whole-body CT, returned normal results. Suspecting an inflammatory process related to SS, high-dose glucocorticoids were initiated.
 Bone scan showing symmetrical hyperenhancement deposits in the distal femoral and tibial diaphysis; B and C) extensive longitudinal transverse myelitis from C3 to C6, without contrast enhancement; D and E) patchy and diffuse hyperintensity in the bulb, pons, and cerebellar peduncles, with areas of patchy intraparenchymal enhancement.")
A) Bone scan showing symmetrical hyperenhancement deposits in the distal femoral and tibial diaphysis; B and C) extensive longitudinal transverse myelitis from C3 to C6, without contrast enhancement; D and E) patchy and diffuse hyperintensity in the bulb, pons, and cerebellar peduncles, with areas of patchy intraparenchymal enhancement.
During treatment, the patient experienced clinical and radiological worsening, accompanied by diabetes insipidus. Further investigation included a bone scintigraphy, which showed symmetrical hyperuptake in the femoral diaphysis and the distal diaphyseal-metaphyseal region of the tibia. A biopsy of the lesions identified foamy histiocytes with positive CD68 immunostaining, consistent with Erdheim-Chester disease (ECD). Molecular studies revealed negativity for the V600E mutation.
After beginning treatment with pegylated interferon alpha (IFN-α), the patient achieved clinical stability. However, she developed ototoxicity secondary to the interferon, requiring a switch to cladribine. She is currently stable under periodic clinical and radiological monitoring.
DiscussionErdheim-Chester disease (ECD) is a rare disorder, with 1500 cases reported in the literature since its description in 1930, although its exact prevalence and incidence remain uncertain. It predominantly affects adults, with an average age at diagnosis of 55 years and a male-to-female ratio of 1.5:1.1
The clinical spectrum is heterogeneous, with disease progression varying depending on the affected organs, ranging from mildly asymptomatic to disseminated and lethal forms. Cardiovascular and central nervous system (CNS) involvement is associated with a worse prognosis.2,3
The most common manifestation, seen in over 95% of patients, is osteosclerosis of the diaphysis and metaphysis of long bones in the lower limbs. Diabetes insipidus, present in up to a third of cases,4,5 is often the first manifestation of the disease. Neurological involvement, observed in more than half of individuals,6 includes intraparenchymal affection, predominantly in the posterior fossa,7 and vascular and meningeal involvement; however, intramedullary infiltration is rare. Notable findings may also include pericardial involvement, circumferential infiltration of the aorta, perirenal fat, and retroperitoneal fibrosis.
The diagnosis of ECD is established through histology and the histiocyte phenotype within the appropriate clinical and radiological context. The diagnostic yield of biopsy varies based on the tissue examined, and multiple attempts are often necessary to achieve confirmation. This, among other factors, contributes to a diagnostic delay averaging between one to 2.7 years.1,4
Biopsy is crucial, but histological findings are not specific to the disease. Immunohistochemical and molecular analysis techniques are essential for accurate diagnosis. Histopathology is characterized by foamy mononuclear histiocytes with small nuclei, multinucleated histiocytes or Touton cells, fibrosis, and the presence of lymphocytes, plasma cells, and reactive neutrophils. In ECD, histiocytes demonstrate positive immunostaining for CD68 and CD163, while being negative for CD1a.8
The identification of mutations in genes associated with the RAS-RAF-MEK-ERK kinase pathway (MAPK pathway) in both LCH and ECD has led to the reclassification of these entities as proliferative neoplasms. Notably, the V600E mutation in the B-rapidly accelerated fibrosarcoma (BRAF) proto-oncogene is present in more than half of EEC cases. Screening for these mutations in clinical practice is beneficial and recommended for confirming challenging diagnoses and for subjects who have not responded to first-line therapies.8
ECD and LCH, both categorized in the “L” group of the current classification of histiocytosis, exhibit differences beyond their clinical presentations. LCH has a higher incidence in pediatric populations and is histologically characterized by pathological mononucleated histiocytes with coffee bean- or kidney-shaped nuclei. It is confirmed by electron microscopy through the identification of cytoplasmic Birbeck granules and shows positive immunostaining for CD1a and CD207.
Despite these observations, the pathogenesis of ECD remains unclear. Evidence suggests that a proinflammatory cytokine pathway may be responsible for the recruitment and activation of pathological histiocytes.1,9 Elevated levels of IFN-α, a regulator of inflammation, along with increased interleukin (IL) 12, monocyte chemotactic protein-1, and decreased levels of IL-4 and IL-7, indicate an underlying systemic immune dysfunction involving T-helper cells.6
It is uncertain whether alterations in these cytokines are a cause or a consequence of pathological changes in histiocytes. However, the surrounding microenvironment, with the recruitment of immune cells, may contribute to the induction of autoimmunity.10 Thus, the observed systemic and local immune activation is proposed not only as a driver of organic damage but may also explain the high prevalence of associated autoimmunity—both clinical and biological—in up to 12% of patient cases, including autoimmune thyroiditis, Sjögren syndrome, and systemic lupus erythematosus.5,11
Most individuals with ECD require treatment, except for those with asymptomatic or mildly symptomatic disease. Various therapeutic regimens, including cytotoxic and immunosuppressive chemotherapies such as vinca alkaloids, anthracyclines, cyclophosphamide, mycophenolate mofetil, and high-dose chemotherapy followed by autologous stem cell transplantation, have been described, although their clinical efficacy is limited. Corticosteroids may provide acute reduction of edema but are not regarded as effective monotherapy.
Treatment options are determined by clinical features and mutational status. First-line therapies typically involve IFN-based regimens, which have shown improved survival rates, although this varies based on organ involvement. Adverse effects such as depression and fatigue may occur, with pegylated forms being better tolerated. Additionally, anakinra, a recombinant IL-1 antagonist, and cladribine have demonstrated moderate efficacy in patient series with clinical and radiological responses.5,11
The discovery of the involvement of the MAPK pathway has enabled the use of targeted therapies such as BRAF inhibitors (vemurafenib and dabrafenib) and MEK inhibitors (cobimetinib and trametinib), primarily in cases with severe disease manifestations or following failure of conventional treatments.2,8 Their use in patient series has demonstrated effectiveness; however, their considerable toxicity underscores the need for close monitoring of treatment-related side effects. Vemurafenib can lead to skin toxicity, cardiac conduction abnormalities, arthralgia, and gastrointestinal issues, while cobimetinib is associated with rhabdomyolysis, retinopathy, and acneiform rash.
Targeted therapies have emerged as a promising alternative in the treatment of ECD. However, the results require careful use of these drugs in appropriate clinical settings, suggesting that further questions remain. Additional studies are needed to better assess the efficacy of combination therapies and to explore the effectiveness of new drugs with improved CNS penetration or alternative mechanisms of action.12
ConclusionErdheim-Chester disease should be considered as a diagnostic possibility in neurological conditions characterized by inflammatory neuroimaging that is refractory to immunomodulatory treatment. Autoimmune disorders are often associated with and may overlap with the clinical presentation of Erdheim-Chester disease. Advances in understanding etiopathogenesis are promising and impact both the diagnosis and treatment of this condition, which, while still rare, is becoming increasingly recognized, leading to more diagnoses and improved expectations for affected individuals.
Ethical considerationsVerbal and written consent was obtained from the patient for the use of clinical data and complementary tests conducted for this purpose.
FinancingThis work has not received any funding.
