



IgG4-related disease is a recently described disease that can involve various organs and systems. Single organ involvement is the exception to the rule, it is generally a multi-system entity. We present a 36-year-old woman, with no previous pathological history or autoimmune disease, with headache caused by cystic macroadenoma. A transsphenoidal resection was performed and pathology documented areas of fibrosis with a predominantly plasmolymphocytic infiltrate and positive IgG4 staining in more than 20 cells per high-power field, meeting diagnostic criteria for IgG4-related sclerosing disease. Involvement of other organs was ruled out, and the patient improved clinically after management.
La enfermedad relacionada con IgG4 es una entidad recientemente descrita, capaz de involucrar diversos órganos y sistemas. El compromiso de órganos aislados es la excepción a la regla, dado que generalmente se trata de una entidad multisistémica. Se presenta el caso de una mujer de 36 años, sin antecedentes patológicos previos, en quien como causa de cefalea se documenta un macroadenoma quístico llevado a resección transesfenoidal, cuyo resultado de patología documenta zonas de fibrosis con infiltrado de predominio plasmolinfocitario y la tinción para IgG4 positiva en más de 20 células por campo de alto poder, lo que configura criterios diagnósticos para enfermedad esclerosante relacionada con IgG4; se descartó compromiso de otros órganos y hubo mejoría clínica posterior al manejo.
Hypophysitis is an exceptional medical condition that presents with inflammation of the pituitary gland, which causes different degrees of hypopituitarism.1 An annual incidence of one case per 9,000,000 people is estimated.2 This disease encompasses a broad clinical spectrum which ranges from manifestations derived from the hormonal deficit to symptoms product of the mass effect resulting from the involvement of local structures.3
IgG4-related disease is a multisystem disorder recently described,4 characterized by fibroinflammatory involvement of different groups of organs, storiform fibrosis and lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells.5,6 Its most common form of presentation includes involvement of the pancreas, bile duct, retroperitoneum, and salivary glands.7–9 However, it can compromise a large number of systems, such as the cardiovascular, pulmonary, renal, integumentary, and nervous.10
Central nervous system manifestations are infrequent, but meningeal, parenchymal, and pituitary involvement has been described.9,11 The case of a young woman with neurological manifestations as the only form of presentation of IgG4-related hypophysitis, confirmed by biopsy and immunohistochemistry is documented in this article.
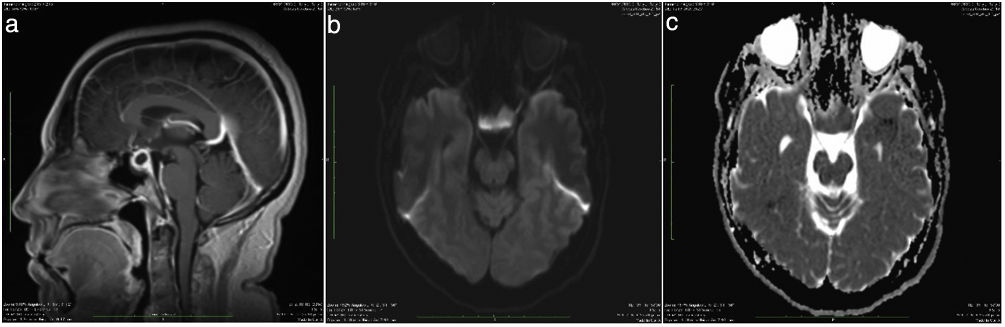
Clinical caseA 36-year-old patient, with no significant personal history, started with a clinical picture of 3 months of evolution consisting of headache predominantly frontal, radiating in a band to the posterior region, episodic, moderate in intensity, which became more frequent over the course of time, with longer duration. Associated with the picture, she began to present diplopia and photophobia. She repeatedly consulted the emergency department due to exacerbation of the symptoms, and there she received symptomatic management, with which the symptoms partially subsided. In the last visit, a contrast-enhanced magnetic resonance (MRI) of the brain was requested, in which a sellar lesion with a suprasellar component of 1.3×1.3×2.4cm was evidenced, with posteriorly displaced infundibulum, without compromise of the optic chiasm, for which a presumptive diagnosis of cystic macroadenoma was established (Fig. 1).
 Intrasellar lesion with suprasellar extension. B) Hypovascular behavior after the application of intravenous contrast. C) The infundibulum and the pituitary gland are displaced posteriorly, exerting an extrinsic compressive effect on the optochiasmatic pathway.")
The patient was referred for evaluation by neurosurgery due to the persistence of the symptoms with the usual analgesic medical treatment (corticosteroids were not used). Liver, thyroid, and pituitary profiles, including basal cortisol, ACTH, prolactin, FSH, LH, estradiol, and IGF1 were requested there, all reported within normal limits (Table 1), therefore, given the lack of diagnostic clarity, it was decided to resect the lesion through endonasal approach for extirpation of the mass and histopathological study, including immunohistochemistry. The surgical procedure was carried out without complications. The pathology study documented areas of fibrosis with a predominantly plasmolymphocytic infiltrate, in addition to positive IgG4 staining in more than 20 cells per high-power field, which constituted diagnostic criteria for IgG4-related sclerosing disease.
Results of the pre-surgical laboratory tests of the patient.
| Diagnostic aids (measurement units) | Results | Normal range |
|---|---|---|
| Creatinine (mg/dl) | 0.9 | 0.51–0.95 |
| LDH (U/l) | 438 | 135–214 |
| ALT (U/l) | 24 | 0–31 |
| AST (U/l) | 28 | 0–32 |
| Fibrinogen (mg/dl) | 53 | 228–415 |
| Leukocytes (×103/μl) | 7.9 | 3.98–10.04 |
| Hemoglobin (g/dl) | 12 | 11.2−15.7 |
| Hematocrit (%) | 32 | 34.1−44.9 |
| Platelets (×103/μl) | 350,000 | 182–369 |
| TSH (μIU/ml) | 3.0 | 0.27–4.2 |
| Free T4 (ng/dl) | 1.5 | 0.93–1.7 |
| Anti-TPO (IU/ml) | 4.0 | <5.61 |
| Basal cortisol (μg/dl) | 11 | 5.0−20 |
| ACTH (pg/ml) | 18 | 9−52 |
| Sodium (mEq/l) | 138 | 136–145 |
| Prolactin (ng/ml) | 24 | 3–25 |
| FSH (follicular phase) (IU/l) | 9 | 2–10 |
| LH (follicular phase) (IU/l) | 6 | 2–6 |
| Estradiol (pg/ml) | 50 | 20−120 |
| IGF1 (adjusted for age and sex) (ng/ml) | 220 | 117−329 |
ACTH: adrenocorticotropic hormone; ALT: alanine aminotransferase; Anti-TPO: anti-thyroid peroxidase antibodies; AST: aspartate aminotransferase; LDH: lactic dehydrogenase; FSH: follicle-stimulating hormone; IGF1: insulin-like growth factor 1; LH: luteinizing hormone; TSH: thyroid stimulating hormone.
In the postoperative follow-up at 3 and 6 months, the pituitary structure by MRI and in turn the IG4 serum values (2.36mg/dl, RV 2.2-201) were normal. However, she continued under multidisciplinary management by rheumatology with corticosteroid of prednisolone type 2.5mg every other day until one year after the clinical picture, when it was finally suspended (November 2018). It must be clarified that there were not pre-surgical IgG4 values.
DiscussionImmunoglobulin G (IgG) is one of the most abundant plasma proteins, accounting for approximately 75% of the humoral antibodies produced by the human being.12 There are 4 subclasses of this immunoglobulin (IgG1, IgG2, IgG3 and IgG4), of which the IgG4 type correspond to the smallest proportion <4%,13 and which has been associated with different allergic diseases and autoimmune disorders.14 Only since the year 2010 the nomenclature “IgG4-related disease” has begun to be generalized15 as a term that describes this entity, capable of involving different organs and systems.16 Historically, it has been described as a condition that mainly involves the pancreas,17–19 however, various publications have described concomitant involvement of the salivary glands, retroperitoneal organs, and even of the lung parenchyma.20 Single organ involvement is the exception to the rule, since IgG4-related disease is mostly a multisystem disease.21 Being a recently described entity, the diagnostic criteria to define were only agreed upon less than a decade ago (Table 2).1,16,22These criteria give preponderance to clinical and imaging findings, as well as to the response to treatment as methods to establish the diagnosis.
Diagnostic criteria for IgG4-related hypophysitis.
| Criterion I — Histopathology of the pituitary gland: mononuclear infiltration of the pituitary gland rich in lymphocytes and plasma cells with >10 IgG4-positive cells per high-power field |
| Criterion II — Pituitary MRI: sellar mass or thickened pituitary stalk |
| Criterion III — Commitment of other organs proven by biopsy: association with IgG4-positive lesions in other organs |
| Criterion IV — Serology: increased serum concentration of IgG4 (>140mg/dl) |
| Criterion V — Response to glucocorticoids: reduction in the size of the pituitary mass and symptomatic improvement with glucocorticoids. |
Hypophysitis can be primary (lymphocytic, autoimmune, granulomatous, xanthomatous, necrotizing, and related to IgG4 disease)23 or secondary (as a result of systemic diseases, immunotherapy, or sellar disease). The case would correspond to the subgroup of IgG4 hypophysitis, a rare condition that involves fibroinflammatory compromise of the pituitary gland,5,24 and that it takes place among the group of non-hormone secreting tumors of the sellar region.22 This characteristic makes it difficult to establish a definitive diagnosis prior to the surgical intervention and pathological analysis of the resected pituitary tissue.25
IgG4-related hypophysitis was first described in 2004,26 but it was only confirmed histopathologically in 200723. At present, 85 cases have been reported in the literature, of which only 38 have confirmation by biopsy and histopathology staining. However, it could be present in 30% of the hypophysitis and may be underdiagnosed.1,27–33 This clinical entity can manifest itself in more than 52% of the cases with panhypopituitarism33 and diabetes insipidus,28,30,34 secondarily to the deficiency of antidiuretic hormone (ADH).33 Only a minor proportion develop compressive symptoms such as headache and visual disturbances.1,10,28,35 In our case, the presentation was a headache of increasing intensity associated with a pituitary cystic macroadenoma without pituitary functional involvement and histological confirmation of IgG4-related disease.
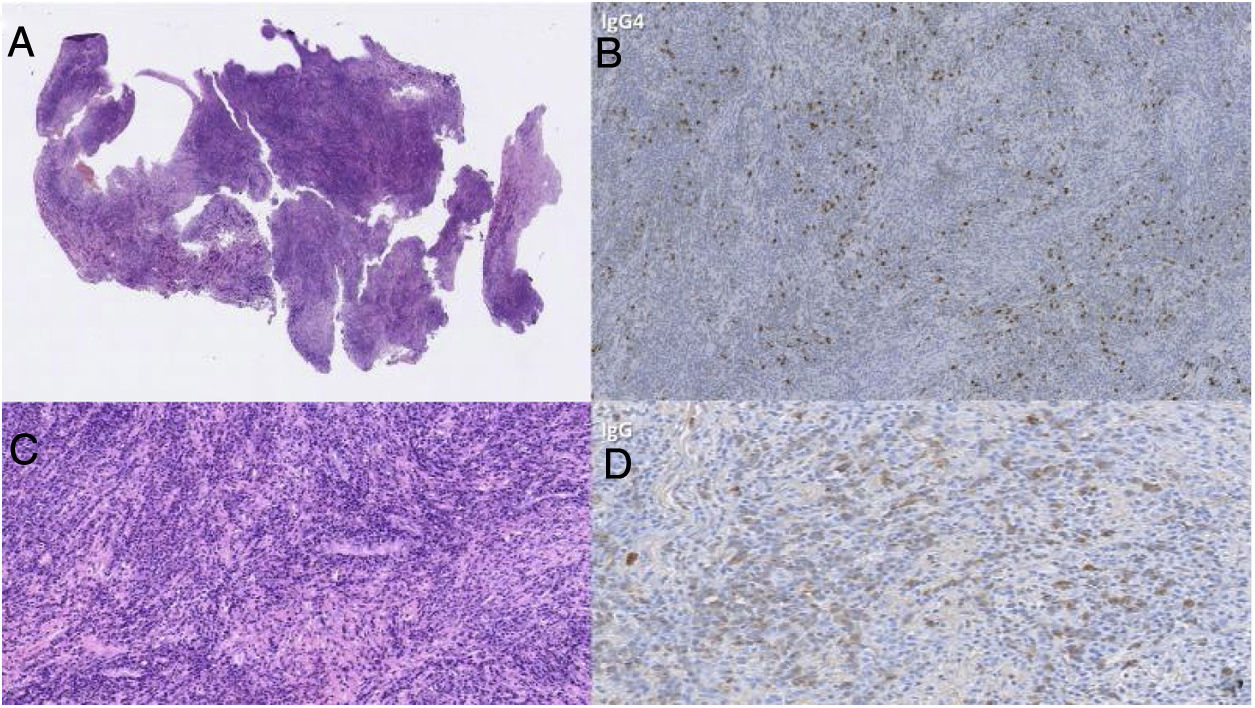
At this moment there is no defined medical therapy for IgG4 hypophysitis, but multiple cases of effectiveness with the use of glucocorticoids as first-line management in patients with IgG4-related disease have been reported.36 On the other hand, in 2013, Hattori et al. described the first case of a patient with a clinical picture of hypophysitis in the absence of pituitary insufficiency who responded satisfactorily to medical management,37 for which they raised the importance of optimizing the diagnostic strategies for this emerging disease; even the response to this type of therapy is part of the diagnostic criteria. Nevertheless, imaging-based diagnosis may be difficult if confirmation is made only by pathology, as in the case presented, in which the H&E staining confirms an abundant lymphoplasmacytic infiltrate within the collagenous stroma, with sclerotic foci, and immunohistochemistry demonstrates an increased IgG4:IgG ratio of 60% (Fig. 2), thus fulfilling the criteria I and II for the diagnosis of IgG4-related hypophysitis.
 H&E staining. Pituitary gland with abundant lymphoplasmacytic infiltrate within the collagenous stroma, with sclerotic foci. B and D) Immunohistochemical technique. Increased IgG4:IgG ratio of 60%.")
Finally, the variability of manifestations presented in the different cohort studies could suggest that there is a genetic predisposition or unknown risk factors that influence the appearance of the disease, without neglecting the importance of the methods used at the time of establishing the diagnosis.20 It is necessary to optimize the diagnostic strategies for this pathology and thus be able to offer optimal therapeutic approaches to the patients.
ConclusionsIgG4-related disease is a recently described entity, capable of involving several organs and systems. A high index of suspicion is important for timely diagnosis and management.
Ethical considerationsThe work was approved by the Biomedical Research Ethics Committee CRI/CE No. 242 - 2020 - Fundación Valle del Cauca. CEII No. 242 - 2020 - Valle del Lili Foundation. Principal Investigator: Dr. Guillermo E. Guzmán. Co-Investigators: Dr. Andrés Hormaza, Dr. Luz Fernanda Sua, Dr. Sergio Ortega, Dr. Daniel Ortiz, Dr. Veline Martínez. Its Good Clinical Practice of Good Clinical Practice have been approved by this Committee in the same review, as well as the informed consent.
Conflict of interestThe authors declare that they have no conflict of interest for the preparation of this article.
