



La enfermedad relacionada con IgG4 es una entidad recientemente descrita, capaz de involucrar diversos órganos y sistemas. El compromiso de órganos aislados es la excepción a la regla, dado que generalmente se trata de una entidad multisistémica. Se presenta el caso de una mujer de 36 años, sin antecedentes patológicos previos, en quien como causa de cefalea se documenta un macroadenoma quístico llevado a resección transesfenoidal, cuyo resultado de patología documenta zonas de fibrosis con infiltrado de predominio plasmolinfocitario y la tinción para IgG4 positiva en más de 20 células por campo de alto poder, lo que configura criterios diagnósticos para enfermedad esclerosante relacionada con IgG4; se descartó compromiso de otros órganos y hubo mejoría clínica posterior al manejo.
IgG4-related disease is a recently described disease that can involve various organs and systems. Single organ involvement is the exception to the rule, it is generally a multi-system entity. We present a 36-year-old woman, with no previous pathological history or autoimmune disease, with headache caused by cystic macroadenoma. A transsphenoidal resection was performed and pathology documented areas of fibrosis with a predominantly plasmolymphocytic infiltrate and positive IgG4 staining in more than 20 cells per high-power field, meeting diagnostic criteria for IgG4-related sclerosing disease. Involvement of other organs was ruled out, and the patient improved clinically after management.
La hipofisitis es una condición médica excepcional que cursa con inflamación de la glándula hipófisis, lo cual ocasiona diferentes grados de hipopituitarismo1. Se estima una incidencia anual de un caso por cada 9.000.000 de personas2. Esta enfermedad comprende un espectro clínico amplio que abarca desde manifestaciones derivadas del déficit hormonal hasta sintomatología producto del efecto masa resultante del compromiso de estructuras locales3.
La enfermedad por IgG4 es un trastorno multisistémico descrito recientemente4, caracterizado por compromiso fibroinflamatorio de diferentes grupos de órganos, fibrosis estoriforme e infiltrado linfoplasmocitario rico en células plasmáticas positivas para IgG45,6. Su forma de presentación más común incluye afección de páncreas, vía biliar, retroperitoneo y glándulas salivares7–9. Sin embargo, puede comprometer un gran número de sistemas, como el cardiovascular, el pulmonar, el renal, el tegumentario y el sistema nervioso10.
Son poco frecuentes las manifestaciones en el sistema nervioso central, pero se ha descrito afección meníngea, parenquimatosa e hipofisiaria9,11. En este artículo se documenta el caso de una mujer joven con manifestaciones neurológicas como única forma de presentación de hipofisitis relacionada con IgG4, confirmada por biopsia e inmunohistoquímica.
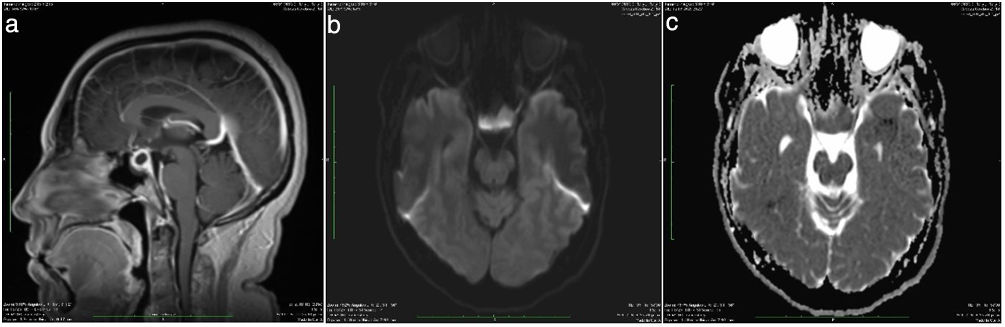
Caso clínicoPaciente de 36 años, sin antecedentes personales de importancia, inició con cuadro de 3 meses de evolución consistente en cefalea de predominio frontal, irradiado en banda hacia región posterior, episódico, intensidad moderada, que en el transcurso de este tiempo se fue haciendo más frecuente, con mayor duración. Asociado al cuadro, empezó a presentar diplopía y fotofobia. Consultó en repetidas ocasiones al servicio de urgencias por exacerbación de los síntomas, y allí le daban manejo sintomático, con lo cual los síntomas cedían parcialmente. En la última atención de urgencia se indicó realización de resonancia magnética nuclear (RMN) de cerebro contrastada, en la que se evidenció lesión selar con componente supraselar de 1,3×1,3×2,4cm, con infundíbulo desplazado hacia posterior, sin compromiso del quiasma óptico, por lo que se hizo diagnóstico presuntivo de macroadenoma quístico (fig. 1).
 Lesión intraselar con extensión supraselar. B) Comportamiento hipovascular posterior a la aplicación de contraste intravenoso. C) El infundíbulo y la glándula hipofisaria se encuentran desplazados posteriormente, ejerciendo efecto compresivo extrínseco sobre la vía optoquiasmática.")
A) Lesión intraselar con extensión supraselar. B) Comportamiento hipovascular posterior a la aplicación de contraste intravenoso. C) El infundíbulo y la glándula hipofisaria se encuentran desplazados posteriormente, ejerciendo efecto compresivo extrínseco sobre la vía optoquiasmática.
La paciente fue remitida a valoración por neurocirugía, debido a la persistencia de la sintomatología con el tratamiento médico analgésico habitual (no se emplearon corticoides). Allí se solicitó perfil hepático, tiroideo e hipofisario, entre lo que se incluía cortisol basal, ACTH, prolactina, FSH, LH, estradiol e IGF1, reportados todos dentro de parámetros normales (tabla 1), por lo cual, ante la falta de claridad diagnóstica, se decidió resección de la lesión por abordaje endonasal para extirpación de la masa y estudio histopatológico, incluyendo inmunohistoquímica. El procedimiento quirúrgico se efectuó sin complicaciones. El estudio de patología documentó zonas de fibrosis con infiltrado de predominio plasmolinfocitario, además de tinción para IgG4 positiva en más de 20 células por campo de alto poder, lo que configuró criterios diagnósticos para enfermedad esclerosante relacionada con IgG4.
Resultado de laboratorios prequirúrgicos de la paciente
| Ayudas diagnósticas (unidades de medición) | Resultados | Rango normal |
|---|---|---|
| Creatinina (mg/dl) | 0,9 | 0,51-0,95 |
| DHL (U/l) | 438 | 135-214 |
| ALT (U/l) | 24 | 0-31 |
| AST (U/l) | 28 | 0-32 |
| Fibrinógeno (mg/dl) | 53 | 228-415 |
| Leucocitos (×103/μl) | 7,9 | 3,98-10,04 |
| Hemoglobina (g/dl) | 12 | 11,2-15,7 |
| Hematocrito (%) | 32 | 34,1-44,9 |
| Plaquetas (×103/μl) | 350.000 | 182-369 |
| TSH (μUI/ml) | 3,0 | 0,27-4,2 |
| T4 libre (ng/dl) | 1,5 | 0,93-1,7 |
| Anti-TPO (UI/ml) | 4,0 | <5,61 |
| Cortisol basal (μg/dl) | 11 | 5,0-20 |
| ACTH (pg/ml) | 18 | 9-52 |
| Sodio (mEq/l) | 138 | 136-145 |
| Prolactina (ng/ml) | 24 | 3-25 |
| FSH (fase folicular) (UI/l) | 9 | 2-10 |
| LH (fase folicular) (UI/l) | 6 | 2-6 |
| Estradiol (pg/ml) | 50 | 20-120 |
| IGF1 (ajustado a edad y sexo) (ng/ml) | 220 | 117-329 |
CTH: hormona adrenocorticotropa; ALT: alanina aminotransferasa; Anti-TPO: anticuerpos antiperoxidasa tiroidea; AST: aspartato aminotransferasa; DHL: deshidrogenasa láctica; FSH: hormona foliculoestimulante; IGF1: factor de crecimiento insulínico tipo 1; LH: hormona luteinizante; TSH: hormona estimulante de tiroides.
En el seguimiento postoperatorio a 3 y 6 meses, la estructura hipofisaria por RMN y a su vez los valores de IG4 séricos (2,36mg/dl VR 2,2-201) fueron normales. Sin embargo, continuó en manejo multidisciplinario por reumatología, con corticoesteroide tipo prednisolona 2,5mg de forma interdiaria hasta un año posterior al cuadro, cuando finalmente fue suspendido (noviembre del 2018). Es de aclarar que no se contó con valores prequirúrgicos de IgG4.
DiscusiónLa inmunoglobulina G (IgG) es una de las proteínas plasmáticas más abundantes, representa aproximadamente el 75% de los anticuerpos humorales producidos por el ser humano12. Existen 4 tipos de subclases de esta inmunoglobulina (IgG1, IgG2, IgG3 e IgG4), de las cuales el tipo IgG4 corresponde a la proporción más escasa <4%13, y la cual se ha asociado con diferentes enfermedades alérgicas y trastornos autoinmunes14. Solo desde el año 2010 se ha empezado a generalizar la nomenclatura «enfermedad relacionada con IgG4»15 como un término que describe esta entidad, capaz de involucrar diversos órganos y sistemas16. Históricamente, se ha descrito como una afección que compromete principalmente páncreas17–19, sin embargo, diversas publicaciones han descrito compromiso concomitante de glándulas salivares, órganos retroperitoneales e incluso del parénquima pulmonar20. El compromiso de órganos aislados es la excepción a la regla, dado que la enfermedad relacionada con IgG4 se trata en su mayor parte de una enfermedad multisistémica21. Al ser una entidad recientemente descrita, los criterios diagnósticos para definirla se concertaron hasta hace menos de una década (tabla 2)1,16,22. Estos criterios dan preponderancia a hallazgos clínicos e imagenológicos, así como a la respuesta al tratamiento como métodos para establecer el diagnóstico.
Criterios de diagnóstico para hipofisitis relacionada con IgG4
| Criterio I - Histopatología hipofisaria: Infiltración mononuclear de la glándula hipófisis rica en linfocitos y células plasmáticas con >10 células positivas para IgG4 por campo de alta potencia |
| Criterio II - RMN hipófisis: Masa selar o tallo hipofisiario engrosado |
| Criterio III - Compromiso comprobado por biopsia en otros órganos: Asociación con lesiones positivas para IgG4 en otros órganos |
| Criterio IV - Serología: Aumento de la concentración sérica de IgG4 (>140mg/dl) |
| Criterio V - Respuesta a glucocorticoides: Disminución del tamaño de la masa hipofisaria y mejoría sintomática con glucocorticoides |
Las hipofisitis pueden ser primarias (linfocitaria, autoinmune, granulomatosa, xantomatosa, necrosante y relacionada con enfermedad por IgG423) o secundaria (como resultados de enfermedades sistémicas, inmunoterapia o enfermedad selar). El caso correspondería al subgrupo de hipofisitis por IgG4, una condición rara que involucra compromiso fibroinflamatorio de la glándula hipófisis5,24, y que toma lugar entre el grupo de tumores de la región selar no secretores de hormona22. Esta característica dificulta el establecimiento de un diagnóstico definitivo previo a la intervención quirúrgica y al análisis patológico del tejido hipofisario resecado25.
La hipofisitis relacionada con enfermedad por IgG4 se describió por primera vez en el 200426, pero solo se confirmó histopatológicamente hasta el 200723. En la actualidad, en la literatura se han reportado 85 casos, de los cuales apenas 38 cuentan con confirmación por biopsia y tinciones de histopatología. Sin embargo, podría estar presente en el 30% de las hipofisitis y ser subdiagnosticada1,27–33. Esta entidad clínica se puede manifestar en más del 52% de los casos con panhipopituitarismo33 y diabetes insípida28,30,34, secundariamente al déficit de hormona antidiurética (ADH)33. Solo una proporción menor desarrolla síntomas compresivos tales como cefalea y trastornos visuales1,10,28,35. En nuestro caso, la presentación fue un cuadro de cefalea de intensidad en ascenso asociada a un macroadenoma quístico hipofisario sin afectación funcional hipofisaria y confirmación histológica de enfermedad relacionada por IgG4.
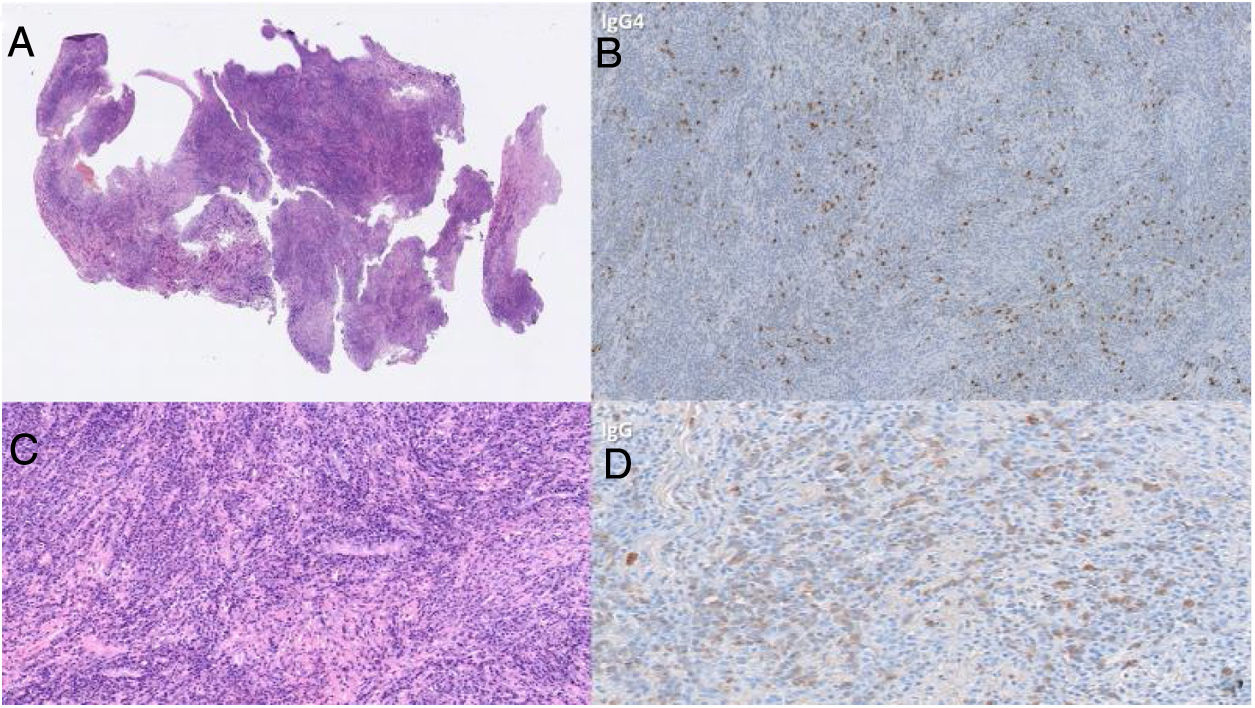
En este momento no existe una terapia médica definida para la hipofisitis por IgG4, pero se han reportado múltiples casos de efectividad en el uso de glucocorticoides como manejo de primera línea en pacientes con enfermedad relacionada con IgG436. Por otro lado, en el 2013, Hattori et al. describieron el primer caso de un paciente con un cuadro de hipofisitis en ausencia de insuficiencia hipofisaria que respondió satisfactoriamente al manejo médico37, por lo que plantearon la importancia de optimizar las estrategias diagnósticas frente a esta enfermedad emergente; incluso la respuesta a este tipo de terapia hace parte de los criterios diagnósticos. No obstante, el diagnóstico por imágenes puede ser difícil si solo se hace confirmación por patología, como en el caso presentado, en el que la coloración de H&E confirma infiltrado linfoplasmocitario abundante dentro del estroma colágeno, con focos escleróticos, y la inmunohistoquímica demuestra una relación incrementada de IgG4:IgG del 60% (fig. 2), cumpliéndose así los criterios I y II para el diagnóstico de hipofisitis relacionada con IgG4.
 Coloración de H&E. Hipófisis con infiltrado linfoplasmocitario abundante dentro del estroma colágeno, con focos escleróticos. B y D) Técnica de inmunohistoquímica. Relación incrementada de IgG4:IgG del 60%.")
Para finalizar, la variabilidad de manifestaciones presentadas en los diferentes estudios de cohortes podría sugerir que existe una predisposición genética o factores de riesgo desconocidos que influyen en la aparición de la enfermedad, sin dejar de lado la importancia de los métodos empleados al momento de realizar el diagnóstico20. Es necesario optimizar las estrategias de diagnóstico para esta patología y poder ofrecer así abordajes terapéuticos óptimos para los pacientes.
ConclusionesLa enfermedad relacionada con IgG4 es una entidad descrita recientemente, capaz de involucrar diversos órganos y sistemas. Un índice de sospecha alto es importante para el diagnóstico y el manejo oportuno.
Consideraciones éticasEl trabajo fue aprobado por el Comité de Ética en Investigación Biomédica IRB/EC No. 242 – 2020 - Fundación Valle del Lili. Investigador Principal: Dr. Guillermo E. Guzmán. CoInvestigadores: Dr. Andrés Hormaza, Dra. Luz Fernanda Sua, Dr. Sergio Ortega, Dr. Daniel Ortiz, Dra. Veline Martínez. Sus certificados de curso Buenas Prácticas Clínicas han sido aprobados por este Comité en la misma revisión, asimismo como el consentimiento informado.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses para la elaboración de este artículo.
