



La pancreatitis autoimmune es una manifestación característica del espectro de la enfermedad relacionada con IgG4, trastorno raro de tipo autoinmune que se presenta clínicamente con ictericia obstructiva debido a la infiltración de células plasmáticas y fibrosis en el páncreas; puede presentarse con otra sintomatología en caso de afectación de otros órganos y en muy raras ocasiones hay compromiso hematológico. Se presenta el caso de un hombre adulto con signos de colestasis secundaria a una pancreatitis autoinmune tipo i, con compromiso de otros órganos y asociada con trombocitopenia que mejoró con el tratamiento inmunosupresor a base de corticoide sistémico, luego del cual se observó una evolución favorable en cuanto a la clínica y analítica en el transcurso del tiempo.
Autoimmune pancreatitis is a characteristic manifestation of the spectrum of the disease related to IgG4, a rare autoimmune disorder that presents clinically with obstructive jaundice due to the infiltration of plasma cells and fibrosis in the pancreas. There may be other symptoms in case of involvement of other organs, and in very rare cases there is haematological involvement. We present the case of an adult man with signs of cholestasis secondary to type I autoimmune pancreatitis, with involvement of other organs and associated with thrombocytopenia that improved with systemic corticosteroid-based immunosuppressive treatment, after which the patient showed favourable clinical and analytical evolution over time.
Hombre de 60 años, sin antecedentes clínicos ni quirúrgicos conocidos quien acudió al servicio de Urgencia por cuadro de 8días de evolución caracterizado por presencia de equimosis y petequias espontáneas en el abdomen que posteriormente se generalizaron al resto del cuerpo; 3días antes del ingreso presentó además ictericia, prurito, acolia y coluria. Como síntoma acompañante refirió pérdida de apetito y peso no cuantificado, sin que presentara dolor abdominal ni alza térmica.
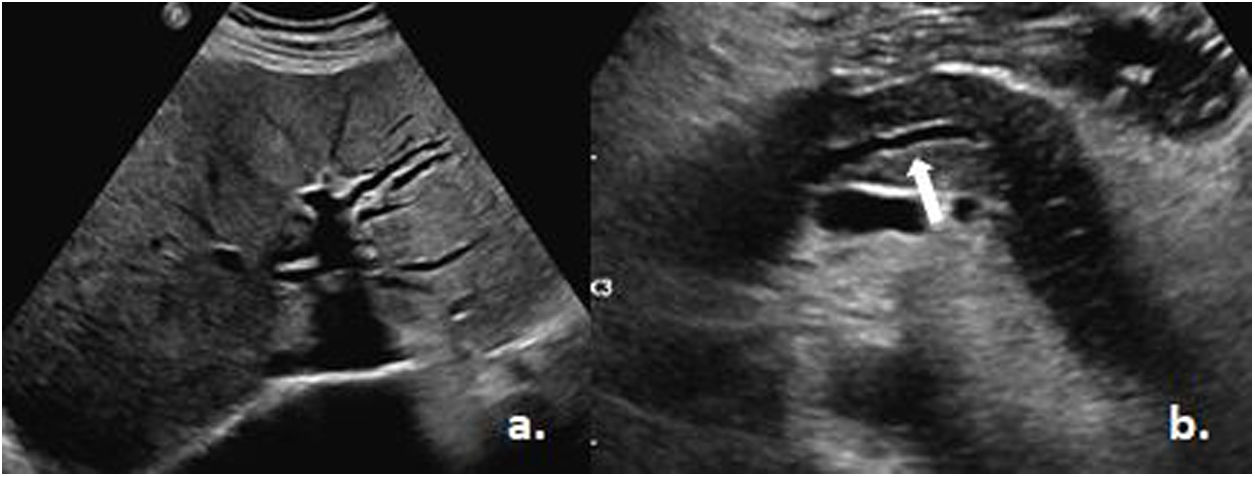
Al ingreso el paciente se encontraba despierto, lúcido, afebril, con signos vitales dentro de los parámetros normales e ictericia generalizada. Dentro de los estudios paraclínicos se encontró leucocitopenia, linfopenia, trombocitopenia severa, así como perturbación en el perfil hepático con presencia de hiperbilirrubinemia a expensas de la directa y elevación de enzimas hepáticas con predominio del patrón colestásico (tabla 1). Se realizó una ecografía de abdomen que no reportó lesiones obstructivas ni ocupativas de espacio, con marcada dilatación de la vía biliar intra y extrahepática y presencia de un páncreas hipoecogénico de tamaño normal con leve dilatación del Wirsung (fig. 1).
Seguimiento bioquímico y hematológico
| Laboratorio evolución | Día 0 | Día 1 | Día 2 | Día 3 | Día 4 | Día 5 | Día 11 | Día 17 | Día 33 | Día 52 | 1.er año |
|---|---|---|---|---|---|---|---|---|---|---|---|
| B. total | 12,9 | 8,07 | 5,63 | 5,31 | 3,8 | 2,1 | 1,04 | ||||
| B. directa | 11,36 | 6,93 | 4,86 | 4,51 | 3,07 | 1,87 | 0,73 | ||||
| B. indirecta | 1,57 | 1,14 | 0,77 | 0,8 | 0,73 | 0,23 | 0,31 | ||||
| GGT | 890 | 528 | 321 | 149 | 50 | ||||||
| FA | 540 | 342 | 267 | 200 | 91 | ||||||
| AST | 132 | 51 | 38 | 33 | 17 | 18 | |||||
| ALT | 160 | 106 | 115 | 93 | 39 | 20 | |||||
| Leucocitos | 3.800 | 3.810 | 3.600 | 4.590 | 8.830 | 7.300 | 7.100 | 11.010 | 6.510 | 9.360 | |
| Linfocitos | 470 | ||||||||||
| Plaquetas | 6.000 | 5.000 | 9.000 | 24.000 | 82.000 | 136.000 | 193.000 | 186.000 | 171.000 | 278.000 | 193.000 |
| IgG4 | 887 (3-201) | 88,7 | |||||||||
| CA 19-9 | 1747 (0-34) | 866,8 | 16,92 |
Valor de: bilirrubinas: mg/dl; GGT, AST, ALT: U/l; leucocitos, linfocitos y plaquetas: K/uL; IgG4: mg/dl; Ca 19-9: U/ml.
Tomado del servicio de la base de datos del Hospital Metropolitano.
 Dilatación de la vía biliar. b) Dilatación del Wirsung (flecha blanca). Tomado del Servicio de Imagenología del Hospital Metropolitano.")
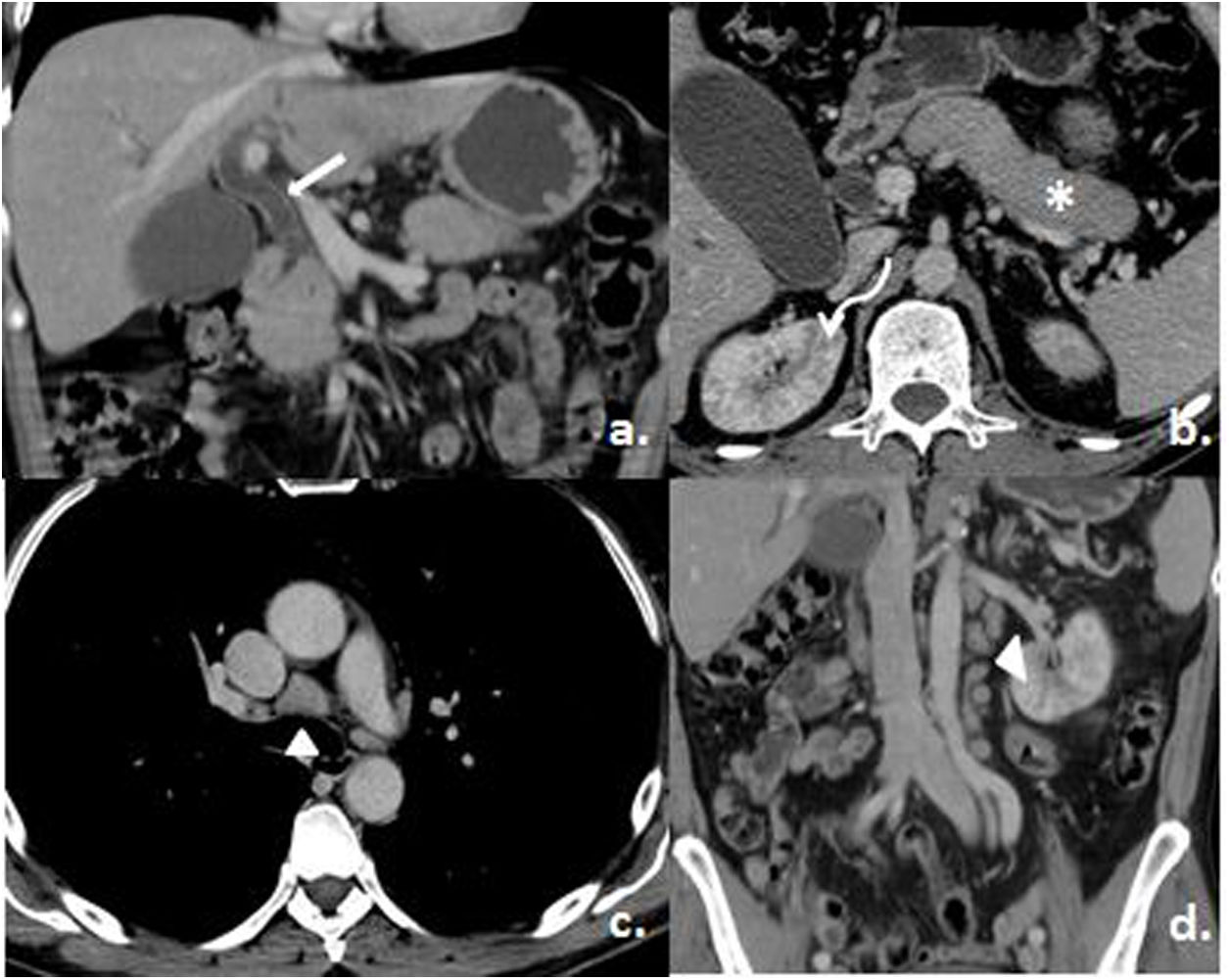
Frente a la dilatación importante de la vía biliar, se solicitó estudio tomográfico que reportó una dilatación del conducto colédoco (16mm), con engrosamiento parietal de toda la vía biliar y refuerzo con el contraste. El páncreas mostraba alteración en su morfología, de forma difusa, con un área más prominente en el proceso uncinado y una lesión focal sospechosa sin identificar. Además, se encontraron múltiples áreas cuneiformes por falta de captación del contraste en ambas corticales de los riñones, de distribución difusa, y múltiples ganglios supraclaviculares, mediastinales (en espacio prevascular y pretraqueal, el de mayor tamaño a este nivel de aproximadamente 2,7×1,9cm), en torno al tronco celíaco, hilio hepático y de mayor tamaño en el retroperitoneo paraaórtico izquierdo y adyacente a la cava, así como en región inguinal e ilíacas bilaterales (fig. 2).
 Dilatación y realce del colédoco (flecha blanca). b) Pérdida de sus lobulaciones periféricas del páncreas (asterisco) y defectos múltiples de la perfusión renal (flecha curva). c y d) Adenomegalias mediastinales y retroperitoneales (puntas de flecha). Tomado del Servicio de Imagenología del Hospital Metropolitano.")
Tomografía contrastada de tórax y abdomen. a) Dilatación y realce del colédoco (flecha blanca). b) Pérdida de sus lobulaciones periféricas del páncreas (asterisco) y defectos múltiples de la perfusión renal (flecha curva). c y d) Adenomegalias mediastinales y retroperitoneales (puntas de flecha).
Tomado del Servicio de Imagenología del Hospital Metropolitano.
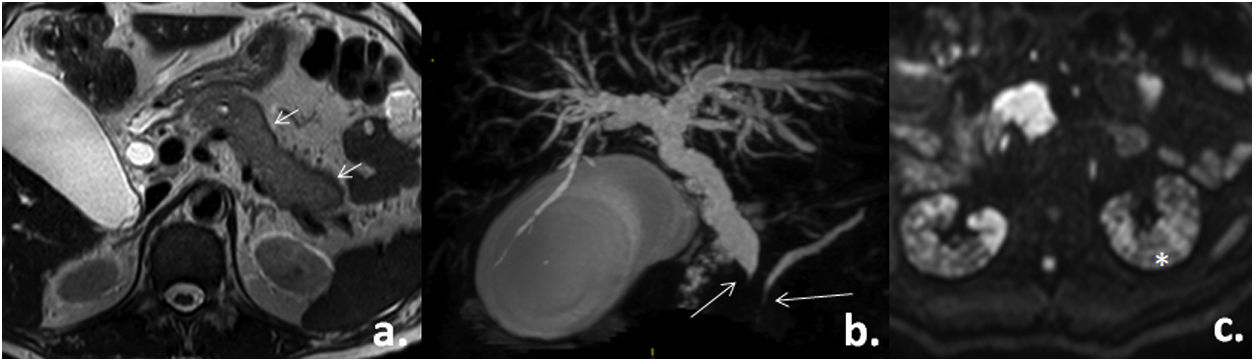
Para una mejor visualización del páncreas se solicitó una resonancia magnética en la que se evidenció la pérdida de la arquitectura pancreática en forma global, con hipointensidad periférica, estrechamiento del colédoco distal con dilatación proximal (14mm de calibre) y del Wirsung distal. Se asocian algunas adenomegalias paraaórticas izquierdas y alteración en parches de ambos riñones (fig. 3).
 Corte axial T2: alteración de la morfología pancreática (flechas cortas). b) Reconstrucción volumétrica de la vía biliar: estrechamiento filiforme del conducto colédoco y del Wirsung con dilatación proximal (flechas largas). C) Corte axial en difusión: alteración en parches de la señal renal (asterisco). Tomado del Servicio de Imagenología del Hospital Metropolitano.")
Resonancia magnética de abdomen. a) Corte axial T2: alteración de la morfología pancreática (flechas cortas). b) Reconstrucción volumétrica de la vía biliar: estrechamiento filiforme del conducto colédoco y del Wirsung con dilatación proximal (flechas largas). C) Corte axial en difusión: alteración en parches de la señal renal (asterisco).
Tomado del Servicio de Imagenología del Hospital Metropolitano.
Frente a un paciente adulto con un cuadro clínico agudo de ictericia por colestasis, trombocitopenia severa, más hallazgos de imagen indicativos de proceso inflamatorio que se asemeja a un seudotumor pancreático que justifica el proceso obstructivo de vía biliar, se sospecharon causas inflamatorias, autoinmunes o neoplásicas; estas últimas menos probables puesto que no se puso de manifiesto una lesión tumoral bien estructurada sin captación de contraste. Se solicitó la cuantificación de IgG total, la cual reportó un valor de 3.183mg/dl (valor normal: 3-201mg/dl), con un IgG4 de 887mg/dl (rango normal: 3-201mg/dl) e IgE 243 UI/ml (normal < 100 UI/ml); además de la medición del marcador tumoral Ca 19-9, que estuvo significativamente elevado (50N), y un antígeno carcinoembrionario que resultó negativo. Se solicitaron también pruebas inmunológicas que reportaron una hipocomplementemia con un C3: 22 pg/ml (normal: 0-8 pg/ml) y C4: 47mg/dl (normal: 90-180mg/dl), así como una elevación de la velocidad de sedimentación por Westergreen de 59mm/1h (rango 3-5) y 73mm/2h (rango 7-15). Los anticuerpos antinucleares fueron positivos, con dilución 1:100 y un estudio de anti-DNA, anticuerpos anticitoplasma de neutrófilos, anticuerpos antimúsculo liso y anticuerpo microsomal renal tipo 1 del hígado, que fueron negativos. Por otro lado, no se afectó la función renal (creatinina 1,0mg/dl); sin embargo, el paciente presentó una oligoalbuminuria cuantitativa de 153,9mg/l (rango 0-20mg/l).
No se llevó a cabo un estudio de glándulas salivales puesto que el paciente no presentó ninguna clínica; en el estudio tomográfico tampoco se reportó ningún hallazgo que sugiriera alguna anormalidad. Así mismo, no se solicitaron estudios para detección de infección por Helicobacter pylori (H. pylori) ni tampoco electroforesis de proteínas e inmunofijación.
Por las manifestaciones clínicas, los biomarcadores y los estudios de imagen realizados, se consideró que el paciente cumplía criterios para una pancreatitis autoinmune tipo i asociada con una trombocitopenia severa. En tal contexto, se inició manejo con metilprednisolona 1 g por vía intravenosa diario por 3días, seguido por prednisona 1mg/kg/día. La evolución clínica del paciente fue satisfactoria, con una disminución progresiva de las petequias, la equimosis y la ictericia que se correlacionó con los datos de laboratorio; estos últimos reportaron una disminución temprana de los marcadores colestásicos y una recuperación de la trombocitopenia hasta llegar a la normalidad a partir del undécimo día de tratamiento, así como una disminución de la IgG4 y del Ca 19-9 (tabla 1).
A la cuarta semana se llevó a cabo una ecografía endoscópica que mostró un parénquima pancreático hiperecogénico de manera difusa y algo nodular. En el transcurso de su evolución el paciente desarrolló diabetes secundaria a la pancreatitis o uso de corticoide, por lo que requirió la instauración de tratamiento a base de insulina.
A los 2años en una ecografía realizada al paciente no se reportaron lesiones focales ni dilatación de vías biliares, con un diámetro del colédoco de 4,2mm; después de 3años el paciente se mantiene con un tratamiento a base de prednisona (5mg/día), asintomático y con exámenes de laboratorio normales.
DiscusiónLa enfermedad relacionada con IgG4 es un desorden autoinmune raro, reconocido por primera vez en el año 2003, caracterizado por la infiltración linfocitaria y de células plasmáticas positivas a IgG4 en casi todos los órganos1, que incluyen: glándulas salivales (submandibulares, partidas y sublinguales) y glándulas lacrimales, las cuales en conjunto conforman la enfermedad de Mikulicz, caracterizada clínicamente por dolor y edema en estas glándulas y que, en ocasiones, se confunde con el síndrome de Sjögren; además, se presenta afectación del páncreas y del árbol biliar, pulmones, riñones, hipófisis, aorta, retroperitoneo, meninges y glándula tiroides (tiroiditis de Riedel)2-4.
Una de las presentaciones más frecuentes es la pancreatitis autoinmune, clínicamente caracterizada por una ictericia obstructiva no dolorosa y en ocasiones con el inicio diabético o síntomas de insuficiencia pancreática. Se describen 2tipos: la tipo ii, que es la manifestación más frecuente de la enfermedad relacionada por IgG4, en el que histológicamente se encuentra un infiltrado linfoplasmático rico en células positivas a IgG4 y fibrosis que envuelve los lóbulos pancreáticos, sus ductos y el tejido adiposo peripancreático o de otros órganos; suele afectar más a los hombres mayores de 50 años, y el tipo ii, que histológicamente presenta una pancreatitis ducto-céntrico idiopático y, a diferencia del tipo i, únicamente tiene afectación en el páncreas; se presenta en pacientes más jóvenes, con la misma proporción en los 2sexos y tiene pocas células positivas a IgG41,2.
La asociación de trombocitopenia con la enfermedad por IgG4 es muy rara, su mecanismo fisiopatogénico es autoinmune. En algunos reportes de casos se ha descrito la asociación con la presencia de anticuerpos en la superficie de la plaqueta en la que se encuentra la glucoproteína iib/iiia o ib/ix/iv4. La respuesta a la corticoterapia generalmente es satisfactoria, sin embargo, en ocasiones es necesario añadir otros inmunosupresores3,5.
Es importante tener en cuenta que los pacientes con una enfermedad relacionada con IgG4 tienen afectación multiorgánica, la cual en muchos casos puede confundirse con algún tipo de malignidad, infecciones u otra condición de tipo inmune. Esta enfermedad relacionada con IgG4 tiene 2características que se pueden confundir con una enfermedad neoplásica de tipo gastrointestinal: la primera es que en el estudio tomográfico la imagen que se visualiza producto del proceso inflamatorio en ocasiones se asemeja a un tumor o masa, por lo que se suele describir como un seudotumor pancreático, pero en realidad no lo es. Y la segunda característica es que en la mayoría de los casos se acompaña de niveles séricos de Ca 19-9 elevados, por lo que se necesita la oportuna diferenciación clínica entre una enfermedad tumoral y una autoinmune para evitar tratamientos o cirugías que no se necesiten2-6.
El diagnóstico de esta enfermedad se basa en una combinación de hallazgos clínicos, serológicos, radiológicos y de patología. En tal sentido, se observa el agrandamiento del órgano afectado, masas o lesiones nodulares, niveles séricos de IgG4>1,35g/l (ocurre en el 55-97% de los casos)6 y hallazgos histopatológicos con>10 IgG4 + células/campos de alta potencia, así como una relación de células positivas con IgG4/IgG>40%7. La enfermedad puede manifestarse de diferentes formas dependiendo del órgano comprometido (11 posibles órganos); en la reciente bibliografía hay un documento publicado en el año 2019 por el American College of Rheumatology y la European League Against Rheumatism que presenta los 8criterios de inclusión y los 32 de exclusión para diagnóstico de una enfermedad relacionada con IgG4, tomando en consideración los hallazgos mencionados previamente, sabiendo que cierto número de pacientes presenta esta enfermedad sin elevación de niveles de IgG4 con histopatología confirmativa, lo que probablemente obedezca al grado de afectación sistémica2.
Dentro de los criterios de exclusión serológicos se mencionan la trombocitopenia y la leucocitopenia, debido a que su presentación, como bien se describe, es inusual y generalmente se relaciona con otras enfermedades, como síndromes mielodisplásicos, neoplasias hematopoyéticas y otras condiciones autoinmunes como el lupus eritematoso sistémico2; sin embargo, el paciente cuyo caso se reporta no requirió estudios complementarios como el aspirado o una biopsia de médula ósea debido a que después de 48 h de iniciado el tratamiento mejoró notablemente hasta llegar a su normalización. Por ello, si bien se mencionan criterios diagnósticos, la enfermedad puede tener presentaciones inusuales como esta, que es importante considerar y no excluir en el momento de la valoración integral del paciente con sospecha de esta entidad.
En muchos casos, se ha hecho el diagnóstico de esta enfermedad con base únicamente en la clínica, la imagen y la serología, puesto que el acceso a la biopsia en muchas ocasiones no es posible2. En nuestro paciente no se realizó confirmación histopatológica ya que había una alta sospecha clínica, así como otros estudios complementarios que confirmaban este diagnóstico.
Por otro lado, si bien se ha descrito la asociación de la hipergammaglobulinemia, así como la infección por H. pylori, con el desarrollo de la enfermedad relacionada con IgG4, es importante mencionar que dentro del algoritmo diagnóstico en nuestro paciente una debilidad fue no haber solicitado estudios para detección del H. pylori ni tampoco electroforesis de proteínas e inmunofijación.
El tratamiento de primera línea recomendado desde hace mucho tiempo son los glucocorticoides, que tienen un efecto antiinflamatorio y hacen que la sintomatología y los parámetros de laboratorio alterados mejoren rápidamente. No hay un protocolo establecido de corticoterapia: no obstante, para la inducción de la remisión se recomienda iniciar con 0,6-1mg/kg de prednisona (30-40mg/día) o algún otro esteroide equivalente durante 2-4 semanas, para luego reducir progresivamente 5mg cada 1-2 semanas, con un control regular de laboratorio y de imagen después de 4semanas, de modo que el tratamiento termine en 3-6 meses según la evolución clínica. También se describe el uso de metilprednisolona, a dosis más altas de 1g/día por 3días, especialmente en casos en que el tratamiento urgente resulte necesario y el riesgo de daño del órgano sea alto. De igual manera, está indicado el uso de 2dosis de rituximab 1g por vía intravenosa en un espacio de 15 días7,8. En este paciente se ha preferido mantenerlo con el uso de corticoterapia, gracias a lo cual ha permanecido estable y sin recurrencias.
Se describe que el 40% de los pacientes puede sufrir una recaída dentro de los primeros 3años desde su diagnóstico, en el mismo órgano afectado u otro sitio anatómico, momento en el cual nuevamente se pueden usar glucocorticoides en asociación con otros inmunosupresores tales como azatioprina, micofenolato mofetilo, metotrexato o mercaptopurina, obteniéndose así una remisión de hasta el 93% en comparación con aquellos que solo usan corticoide, el 79% a los 6meses. En aquellos en quienes la respuesta al tratamiento descrito es refractaria, se han utilizado otros agentes inmunosupresores como el rituximab, con una duración más prolongada7,8.
Recientemente, se han desarrollado o aplicado medicamentos que pueden actuar de acuerdo con el mecanismo fisiopatológico de esta enfermedad, que cada día se conoce más, teniendo en cuenta que en la primera fase, netamente inflamatoria, se liberan citocinas como la interleucina 1 (IL-1), la IL-6 y el interferón gamma, entre otras, que se encargarán de activar los linfocitos. De ahí el uso de medicamentos como tocilizuman o anakinra. De esta manera, se pueden evitar los efectos adversos producto de la corticoterapia prolongada y en algunos casos sin tener el efecto deseado por dosis subterapéuticas7.
ConclusiónLa enfermedad relacionada con IgG4 es una enfermedad rara, de etiología autoinmune, con manifestaciones clínicas que dependen del órgano comprometido en cada paciente. Como la presentación más frecuente se observa la pancreatitis autoinmune tipo i, en ocasiones acompañada de afectación glandular salivar y lacrimal, así como otras manifestaciones en diferentes órganos producto de la infiltración plasmocítica, como lo son las aquí descritas. Además, la asociación con la trombocitopenia autoinmune es muy rara, por lo que es importante identificar esta enfermedad y tratarla a tiempo, para evitar que los órganos afectados lleguen al estado en el que se presenta fibrosis y el daño es irreversible, a pesar del tratamiento instaurado, lo que puede conllevar fallo orgánico y la muerte.
Desde el punto de vista clínico, este caso fue de difícil diagnóstico, debido a la poca prevalencia en Colombia y sin otros hallazgos clínicos al ingreso que la ictericia en asociación con la trombocitopenia manifestada clínicamente con equimosis y petequias. Un estudio de imagen demostró un «seudotumor pancreático» con obstrucción de la vía biliar, y ante la sospecha de una enfermedad de tipo inflamatorio se solicitaron estudios complementarios para confirmar una pancreatitis autoinmune.
Los autores de este escrito consideramos que el espectro autoinmune propio de esta enfermedad es todavía un área en desarrollo, que requiere el aporte de equipos multidisciplinarios de investigación que involucren a reumatólogos, internistas, gastroenterólogos, nefrólogos, neumólogos, neurólogos, radiólogos y patólogos, y que estudios de este tipo de enfermedades son trascendentales en el momento del diagnóstico y el manejo de los pacientes.
Consideraciones éticasEl consentimiento informado fue debidamente autorizado por el paciente, con respeto por el derecho a la privacidad. Se recibió aval por parte del Comité de Ética del Hospital Metro-politano para su publicación en revista científica.
FinanciaciónNinguna.
Conflicto de interesesLos autores no declaran conflicto de intereses para la elaboración de este artículo.
Agradecemos a Ángela León Cáceres, magíster en Salud Pública, por su colaboración en la revisión y traducción del artículo, así como al Hospital Metropolitano por permitir el acceso a los datos de historia clínica y laboratorio del paciente.
