



La neuromielitis óptica (NMO), también llamada espectro de la neuromielitis óptica (neuromyelitis optica spectrum disorders) se reconoce como una enfermedad inflamatoria, autoinmune, desmielinizante del sistema nervioso central, mediada por autoanticuerpos contra el receptor de acuaporina 4 (AQP4-IgG) que afecta predominantemente a los nervios ópticos y la médula espinal1-3. Es conocido que los pacientes con trastornos inmunitarios tienen más probabilidades de presentar otras enfermedades autoinmunes; sin embargo, no está completamente descrita la asociación entre artritis idiopática juvenil y NMO5. En este escrito se reporta el caso de una paciente que cursa con artritis idiopática juvenil, inició con compromiso neurológico rápidamente progresivo, y es tratada con medicamentos biológicos1-4.
Optic neuromyelitis (ONM), also called neuromyelitis optica spectrum (Neuromyelitis Optica Spectrum Disorders, NMOSD) is recognised as an inflammatory autoimmune demyelinating disease of the central nervous system, mediated by autoantibodies against the aquaporin-4 receptor (AQP4-IgG). It predominantly affects the optic nerves and the spinal cord. It is known that patients with immune disorders are more likely to present other autoimmune diseases, but the relation between juvenile idiopathic arthritis and ONM has not been completely described. In this paper, we report a case of a patient with juvenile idiopathic arthritis, presenting with a rapidly progressive neurological condition, who is treated with biological drugs.
La neuromielitis óptica (NMO), inicialmente conocida como enfermedad de Devic o síndrome de Devic, constituye una enfermedad desmielinizante inflamatoria autoinmune del sistema nervioso central (SNC) que afecta predominantemente a los nervios ópticos y la médula espinal. Aunque Thomas Clifford Allbutt describió en 1870 la asociación entre el trastorno del nervio óptico unilateral y la mielitis, fue el científico francés Eugene Devic quien utilizó por primera vez el término de «neuromielitis óptica», siendo clasificada inicialmente y hasta hace menos de una década como una variante de la esclerosis múltiple (EM) con compromiso del nervio óptico4; sin embargo, hoy se sabe que es una entidad distinta5.
En el año 2015 se publicaron los nuevos criterios diagnósticos para esta enfermedad6, los cuales resaltan la importancia de la positividad de los autoanticuerpos séricos dirigidos al canal de acuaporina 4 (AQP4-IgG), acompañados de las manifestaciones clínicas y las lesiones observadas en imágenes de resonancia magnética (RM), para la realización del diagnóstico de forma temprana, lo cual constituye un factor determinante en el pronóstico del paciente7-10. La comorbilidad de la NMO con otras enfermedades autoinmunes ha sido considerada un factor de mal pronóstico; sin embargo, la presencia de AQP4-IgG por sí sola no respalda esta asociación. Se ha relacionado con diferentes procesos de tipo autoinmune, como el lupus eritematoso sistémico (LES), el síndrome de Sjögren (SS), la miastenia gravis (MG) y la vasculitis, entre otros. Más recientemente, se ha descrito su asociación con artritis reumatoide (AR)11.
El propósito de este escrito es documentar un caso de NMO en una paciente con antecedente de artritis idiopática juvenil tratada previamente con múltiples fármacos biológicos1,12-14.
Presentación del caso clínicoPaciente femenina de 45 años con diagnóstico previo de artritis idiopática juvenil de 30 años de evolución, tratada con cloroquina, azatioprina y prednisolona. Con anterioridad, ha recibido metotrexato, prednisolona, etanercept, adalimumab, leflunomida y tocilizumab, con los cuales no se observó mejoría de los síntomas, y además presentó efectos adversos descritos por la paciente, como rash cutáneo, somnolencia, inapetencia y alopecia.
Ingresa en el servicio de Urgencias por empeoramiento del cuadro clínico iniciado 10 meses atrás con presencia de paresia del miembro inferior derecho. En la actualidad, consulta por paraparesia y pérdida del control de esfínteres con nivel sensitivo T8. Se realiza diagnóstico de mielitis longitudinalmente extensa por compromiso de más de 3segmentos vertebrales, puesto en evidencia en la RM, por lo cual se indica hospitalizar para hacer estudios de extensión e inicio de metilprednisolona 1g IV cada 24 h por 5 días.
Se procede a practicar una punción lumbar que arroja resultados dentro de la normalidad, resaltando la ausencia de bandas oligoclonales en el líquido cefalorraquídeo. Así mismo, se lleva a cabo una electromiografía de las 4extremidades y se encuentra como hallazgo incidental compromiso de medianos sensitivos con patrón mielínico. Además, se reportan anticuerpos antinucleares (ANA), anti-DNA y complemento, perfil tiroideo y niveles de vitamina B12 dentro de parámetros normales (tabla 1).
Reporte de paraclínicos relevantes realizados a la paciente
| Paraclínico | Resultado |
|---|---|
| PCR | 96 |
| VSG | 38 mm/h |
| Factor reumatoideo | 120 UI/ml |
| Hierro sérico | 81 μg/dl (normal) |
| Niveles de vitamina B12 | 459 pg/dl |
| C3 | 115 mg/dl |
| C4 | 20,6 mg/dl |
| Proteínas totales | 4,5 g/dl |
| Albumina | 3 mg/dl |
| Globulina | 1,5 |
| Relación A/G | 2 |
| T3 libre | 3,8 (normal) |
| T4 libre | 0,97 ng/dl (normal) |
| TSH | 1,0 μUI/ml (normal) |
| Anti-DNA | Negativo |
| ANA | Negativos |
| Anticuerpo contra virus de Epstein-Barr | Negativo |
| AgsHb | Negativo |
| Anticuerpo contra hepatitis C | Negativo |
| Anticuerpos acuaporina 4 | 59,4 (positivo) |
| Calcio iónico | 1,3 mmol/l (normal) |
| Fósforo inorgánico | 1,8 mg/dl (bajo) |
| Electroforesis de proteínas en LCR | No presentó bandas oligoclonales |
| Proteínas en orina al azar | 60 mg/dl (elevadas) |
| Anticuerpos anti-RNP | 7,2 UI (negativo) |
| Anticuerpos anti-Ro | 9,1 UI (negativo) |
| VDRL | No reactivo |
| VIH | Negativo |
| Baciloscopia | Negativa |
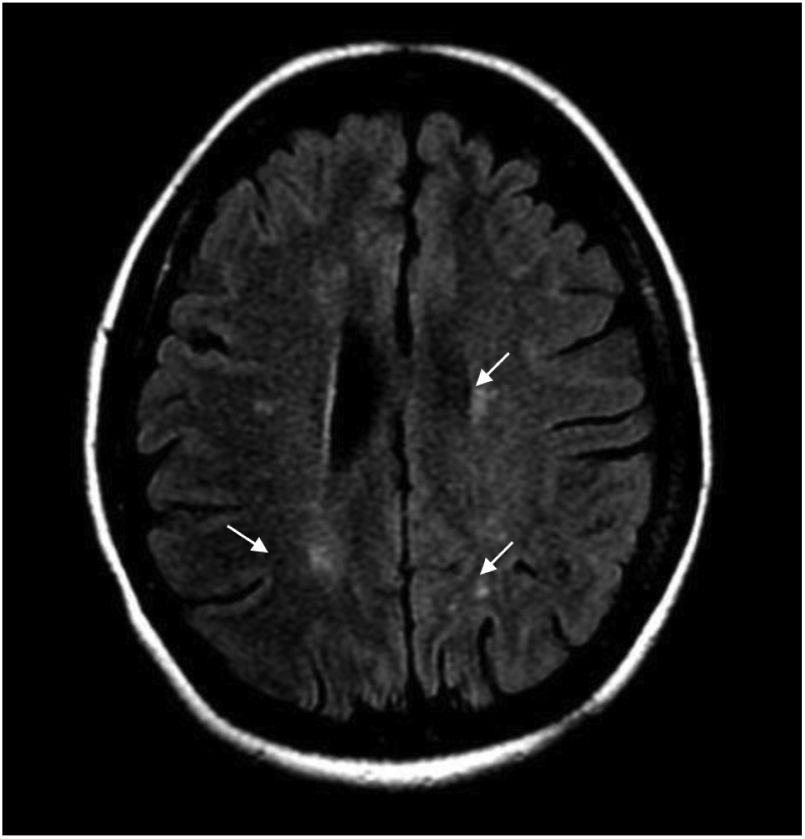
Durante la hospitalización, la paciente evoluciona tórpidamente, presenta dolor neuropático y el nivel sensitivo empeora hasta T6. Se realiza IRM cerebral contrastada y se hallan múltiples imágenes focales paraventriculares y corticosubcorticales con hiperintensidad en secuencia FLAIR, sin realce posterior a la administración del medio de contraste IV ni restricción en secuencias de difusión (fig. 1).
.")
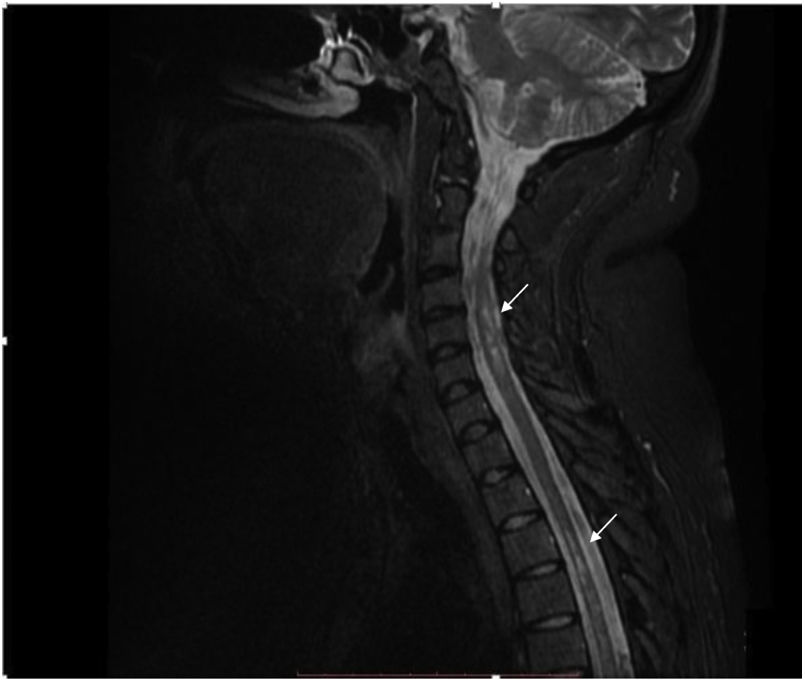
La IRM de columna contrastada muestra imágenes focales hiperintensas a nivel del cordón medular, de ubicación predominantemente lateral y distribución difusa en la columna cervical y dorsal en secuencia T2 (fig. 2). Se considera el diagnóstico de mielitis transversa longitudinalmente extensa en paciente con antecedente de artritis idiopática juvenil.
.")
La paciente fue valorada por los servicios de Medicina Interna, Reumatología y Neurología, y este último solicita niveles séricos AQP4-IgG, los cuales se encuentran elevados (59,4 U/ml), lo que permite hacer el diagnóstico de NMO. Por consiguiente, se decide hacer junta médica para la instauración del tratamiento de la paciente, y Reumatología y Neurología están de acuerdo en iniciar terapia biológica con rituximab, debido al antecedente de uso de múltiples biológicos y anti-TNF sin obtener buena respuesta, puesto que la paciente, aparte de las manifestaciones neurológicas ya descritas, presentaba desviación cubital de las articulaciones metacarpofalángicas, deformidad en cuello de cisne y dolor intenso en articulaciones referidas.
Se inició manejo con rituximab de la siguiente manera: 2 dosis de 1g, separadas por un intervalo de 15 días, y posteriormente 1g cada 6 meses. Durante el seguimiento a un año no se observan nuevas recaídas, no obstante, la paciente quedó con secuelas importantes que la volvieron completamente dependiente para las actividades de la vida diaria.
DiscusiónSe ha reconocido el origen autoinmune de la NMO; esta es una enfermedad de naturaleza desmielinizante e inflamatoria bastante compleja, que presenta una interacción entre factores genéticos y ambientales10. Sigue un curso de recaída en más del 80-90% de los casos, pero su incidencia no está claramente establecida debido al diagnóstico erróneo como EM. Es más prevalente en no caucásicos y en el sexo femenino, con una relación 9:1 en este segundo caso. La edad de inicio varía desde la infancia hasta la edad adulta y afecta principalmente a adultos jóvenes, con una edad media en los Estados Unidos de 41,1 años, a diferencia de la edad de inicio de la EM que generalmente es 10 años antes1,7,14.
Diversos estudios presentan una incidencia que varía de 0,053 a 0,4 por 100.000 individuos y tasas de prevalencia que oscilan entre 0,52 y 4,4 por 100.000 individuos, lo cual la constituye en una enfermedad huérfana13. No obstante, la búsqueda de un diagnóstico con mayor grado de certeza en el menor tiempo posible ha llevado a los expertos a desarrollar los nuevos criterios de NMO.
Con anterioridad al consenso del 2015 se consideraba que la NMO era una enfermedad con un curso monofásico que requería el compromiso del nervio óptico y la médula espinal; sin embargo, con las descripciones posteriores se encontraron nuevos hallazgos de RM que pusieron de manifiesto el compromiso del SNC, el cual puede ser más restringido o más extenso que el demostrado en el nervio óptico y la médula espinal. Aunado a estos hallazgos imagenológicos, se produjo el descubrimiento de los anticuerpos séricos detectables, que se dirigen al canal de AQP4, los cuales están positivos en la mayoría de los pacientes con NMO.
De esta forma, en el 2007 se introdujo el término de «trastornos del espectro NMO» (neuromyelitis optica spectrum disorders [NMOSD]), con lo que se buscaba incluir a aquellos pacientes seropositivos a AQP4-IgG, con formas limitadas o inaugurales de NMO, que tenían un alto riesgo de ataques futuros. También se incluyó a los pacientes con lesiones atípicas de NMO (cerebrales, diencefálicas y del tronco encefálico) y a los que tenían trastornos autoinmunes simultáneos, como es el caso de la paciente que se reporta, sin dejar de lado a aquellos que presentaban EM opticoespinal. Con los criterios del 2015 se unificaron los términos de NMO y NMOSD, dada la incertidumbre diagnóstica y la posible heterogeneidad de la NMOSD seronegativa, dividiendo así el espectro de la enfermedad en aquellos con NMOSD con AQP4-IgG y aquellos con NMOSD sin AQP4-IgG, lo cual permitió que solamente se necesitara tener una característica clínica compatible para realizar el diagnóstico6,15.
En el caso que se reporta aquí, la sospecha diagnóstica se confirmó al asociar las manifestaciones clínicas de la mielitis transversa longitudinalmente extensa, títulos altos del anticuerpo específico y lesiones cerebrales características descritas previamente. Cabe resaltar que sigue siendo un desafío el diagnóstico de este tipo de enfermedades; sin embargo, la presencia de los AQP4-IgG permitió confirmar el diagnóstico, teniendo presente que estos se encuentran hasta en el 75% de los pacientes con NMOSD3,16,17.
En Colombia existen datos de caracterización con los nuevos criterios, según el Consenso Internacional del 2015, que reportan hallazgos similares a otras poblaciones tanto en la clínica como en los datos de laboratorio e imágenes. En el 2016 se publicó un estudio que evaluó una corte de 22 pacientes, en su mayoría mujeres (86%), con una edad promedio de inicio de la enfermedad de 31 años. Se encontró que ninguno de estos pacientes tenía comorbilidad alguna, aunque algunos tuvieron valores positivos de anti-DNA, ANCA, anti-Ro y ANA, por lo que no se descartó la posibilidad de la asociación con otros procesos autoinmunes18.
Aquí se reporta un caso que confirma la posibilidad de que se presenten estas 2patologías autoinmunes asociadas. Se debe destacar que si bien se reconoce que un resultado positivo de ANA no es patognomónico de una enfermedad particular, puede ser de utilidad y debe interpretarse en el contexto de la presentación clínica. También se pueden encontrar títulos o concentraciones bajas de ANA en personas “normales”, a veces en forma transitoria, sobre todo en mujeres mayores de 65 años19.
En la literatura actual se corrobora que existe una asociación fuerte entre NMO y otras enfermedades autoinmunes sistémicas, sin embargo, los datos en relación propiamente con artritis idiopática juvenil y NMO son limitados, lo cual distingue la presentación de nuestra paciente, quien también tuvo un curso rápidamente progresivo11,14.
Hasta la fecha se han notificado diversas enfermedades autoinmunes en aproximadamente el 30% de los pacientes con NMO. Entre las principales se encuentran el LES, el SS, la MG, el síndrome antifosfolipídico, las enfermedades asociadas con ANCA, la tiroiditis de Hashimoto, la anemia perniciosa, la púrpura trombocitopénica idiopática, la colangitis esclerosaste primaria y la sarcoidosis. Lo anterior sugiere una predisposición genética a la poliautoinmunidad, pero faltan más estudios que pongan en evidencia y validen la asociación entre NMO y AR, aunque hoy se conoce que en ambas entidades se presenta una alteración de la inmunidad humoral8,13,20-23.
Recientemente, se ha descrito la importancia de la inmunidad humoral mediada por células B en la patogénesis de la NMO, por lo tanto, una vez se hace el diagnóstico de NMO, la terapia inmunosupresora se debe iniciar lo más pronto posible, buscando retrasar el tiempo de recaída, reducir la gravedad de las recurrencias futuras y minimizar la discapacidad permanente. Existen varios agentes inmunosupresores que han sido utilizados en el tratamiento de la NMO, como el rituximab, el micofenolato, la azatioprina y la mitoxantrona25. Entre estos, el rituximab es la opción terapéutica de elección en pacientes con enfermedades autoinmunes, puesto que se trata de una terapia segura y eficaz tanto en la AR como en la NMO; en Colombia, se ha usado ampliamente con buenos resultados18. Es reconocido por ser el primer anticuerpo monoclonal quimérico de ratón-humano específico para el antígeno CD20 en los linfocitos B que ejerce control de estos mediante la depleción de las células B por citotoxicidad.
El rituximab ha demostrado que disminuye la frecuencia y la severidad de las recaídas en pacientes con NMO. Por otro lado, este anti-CD20 produce mejoría clínica de pacientes con AR25, lo cual se sustenta en diversos ensayos clínicos desde 1998, año en el cual se informó por primera vez la eficacia de rituximab en la AR, reconociendo su efectividad en pacientes en los cuales los inhibidores del TNF han fallado24. De esta, forma se convierte en el medicamento más adecuado para nuestra paciente, quien había presentado reacciones secundarias a otros medicamentos de primera línea, y además se necesitaba disminuir la posibilidad de recaída, ya que la discapacidad secuelar era grave en su caso. Se conoce que el rituximab en pacientes con NMOSD ha demostrado una reducción marcada y sostenida de la tasa de recaída anual, con independencia de los regímenes de inducción y mantenimiento25.
ConclusionesLa fuerte asociación entre NMO y otras enfermedades autoinmunes sistémicas ya ha sido descrita, sin embargo, su concomitancia con la AR aún se conoce poco, por lo cual es importante seguir describiendo reportes de casos como el presentado, en pro de una mejor comprensión de posibles factores de riesgo, fisiopatología y tratamiento en este tipo de enfermedades, en las cuales la poliautoinmunidad es una característica. Se resaltan la eficacia y la seguridad que ha mostrado el advenimiento de las terapias biológicas en estos casos, puntualmente de rituximab, con indicación tanto para la AR como para la NMO.
Consideraciones éticasLa paciente autorizó el uso de su historia clínica para publicación.
Conflicto de interesesLos autores declaran no tener conflicto de intereses para la elaboración de este escrito.
