



TAFRO syndrome is a very rare disease, with less than 100 cases reported in the literature. It is classified as a type of idiopathic multicentric Castleman disease, but it has clinical, paraclinical, and histopathological characteristics that differentiate between TAFRO and idiopathic forms of Castleman disease not otherwise specified. However, it is a challenging exclusion diagnosis. TAFRO syndrome is characterized by systemic inflammatory involvement, often severe, which can present with kidney failure, and become a severe disease with a high mortality rate. The clinical manifestations of TAFRO can be confused with hematology malignancies or various autoimmune diseases. Although there are some reports of TAFRO syndrome associated with autoimmune compromise, there is no published consensus for the diagnosis or treatment. The case presented is a patient who meets the criteria to be classified as SLE, and with manifestations with significant clinical involvement, but with no improvement with standard treatment. It was found that the patient's systemic involvement was due to TAFRO, and that therefore the TAFRO syndrome could simulate SLE, something previously not described in the literature.
El síndrome TAFRO es una enfermedad muy poco común, con menos de 100 casos reportados en la literatura. Se clasifica como un tipo de enfermedad de Castleman multicéntrica idiopática, pero tiene características clínicas, paraclínicas e histopatológicas que permiten diferenciarla de las formas de la enfermedad Castleman idiopática no clasificadas de otra manera; sin embargo, es un diagnóstico de exclusión difícil de hacer. El síndrome TAFRO se caracteriza por compromiso inflamatorio sistémico, en muchas ocasiones severo, que puede presentarse con falla renal y convertirse en una enfermedad grave, con una alta tasa de mortalidad. Las manifestaciones clínicas de TAFRO pueden confundirse con neoplasias hematológicas o varias enfermedades autoinmunes. En la literatura existen algunos reportes de síndrome TAFRO asociados con compromiso autoinmune, pero no se ha publicado un consenso para su diagnóstico ni para su tratamiento. El caso que se presenta es un paciente que cumple con los criterios para ser clasificado como LES, que tenía manifestaciones con gran compromiso clínico, pero sin mejoría con el tratamiento estándar. Se encontró que el compromiso sistémico del paciente era por TAFRO y que, por lo tanto, el síndrome TAFRO podría simular LES, algo no descrito previamente en la literatura.
TAFRO syndrome, also known as Castleman–Kojima disease, is an extremely rare lymphoproliferative disease characterized by multisystemic inflammatory compromise, hence its acronym for thrombocytopenia (T), anasarca (A), fever (F), reticulin fibrosis in bone marrow (R), and organomegaly (O).1 It was first described in 2010 in Japan by Takai et al. as an atypical variant of multicenter Castleman disease (MCD).2 Similar cases have subsequently been reported in Japan,3–8 and in 2014 the first case in a Caucasian patient was reported associated with Sjögren's syndrome.9 In all cases reported of TAFRO syndrome, histological lymph node biopsy studies were conducted, with findings consistent with MCD in patients who were not shown to be human herpesvirus-8 (HHV-8) infection, characterized by lymphoid follicles with atrophic germinal center, large nucleus endothelial cells, endothelial venules proliferation, and a small number of mature plasma cells.10 In 2015, experts met to discuss the classification and diagnostic criteria from 28 previously reported cases,11 with a final update of these diagnostic criteria in 2019.12
TAFRO syndrome differs from other variants of MCD, TAFRO presents lower-grade lymphadenopathy (<1.5cm), normal gamma globulins levels or mild polyclonal hypergammaglobulinemia, and is not associated with HHV-8 or human immunodeficiency virus (HIV), which makes a difference to the classic presentation of MCD associated with these viruses, and that is why it is classified within the group of idiopathic multicenter Castleman disease (iMCD).1,10,12–14 However, there are differences between TAFRO findings and iMCD-not otherwise specified (iMCD-NOS), as iMCD-NOS lymph node pathologies exhibit a pattern with hyperplastic germ centers and linear aggregates of mature plasma cells, unlike the characteristic pathology of TAFRO, which is hypervascular or hyaline-vascular with atrophic germinal centers. Besides, in iMCD-NOS hypogammaglobulinemia is expected, unlike TAFRO where it is mild or absent. Another difference could be the fundamental role of interleukin 6 (IL-6) in iMCD-NOS, which in the case of TAFRO, has been postulated with a less critical role.13–17 Fajgenbaum published that cytokine storm explains the physiopathology in iMCD and proposed different mechanisms, like autoantibodies that stimulate antigenic presentation (autoimmune mechanism), genetic mutations of innate immunity germinal lines (auto-inflammatory mechanism), ectopic cytokines secretion (paraneoplastic mechanism), or signaling by a virus different than HHV-8 (viral mechanism).18
TAFRO syndrome has been reported in middle-aged adults between 43 and 65 years.1,10,14,19 The clinical spectrum is broad and characterized mainly by systemic inflammation manifestations such as fever and fatigue, associated with small lymphadenopathy, and capillary leak syndrome with anasarca and ascites.11–14 A third of patients may have abdominal pain, and less than 15% painful lymphadenopathies. In Iwaki's 2016 publication, which included two American patients with organomegaly, the time from onset of symptoms to biopsy diagnosis was six weeks on average. Laboratory findings were typically severe thrombocytopenia, microcytic anemia, hypoalbuminemia, elevated C-reactive protein (CRP), and high alkaline phosphatase. Bone marrow aspiration and biopsy (BMAB) show plasmacytosis-free myelofibrosis in 80% of cases.10 Mortality associated with TAFRO syndrome is estimated at around 25%. In general, a poor prognosis of patients with iMCD is associated with severe overall commitment to quality of life, hemoglobin levels below 8g/dL, renal failure with estimated glomerular filtration rate (GFR) less than 30mL/min/1.73m2, and the presence of pulmonary commitment, anasarca, ascites or effusions.14,20
Systemic lupus erythematosus (SLE) is associated with the production of multiple autoantibodies. It has a wide variety of clinical manifestations, such as hematological, mucocutaneous, neurological, or renal. SLE's pathological basis is related to multiple autoantibodies that stimulate complement activity resulting in the formation and deposit of immune complexes, in addition to other autoimmune processes such as increased production of some cytokines.21 The variety in the clinical presentation and pathogenesis makes SLE a disease challenging to define, that complexity is why the classification criteria are essential for classifying patients into relatively homogeneous groups. The last update of these classification criteria was published in 201922 where the presence of antinuclear antibodies (ANA) is necessary in all cases to classify patients within SLE, and alterations such as lupus nephropathy, double-chain anti-DNA antibodies (anti-DNA) or complement levels also help define the disease.23 Typical cases of SLE can be diagnosed relatively easily without any doubt, but in some cases, only a few characteristics are present, and the findings may resemble other autoimmune, infectious, or hematological diseases.24 Some patients may have a combination of SLE clinical findings and even meet classification criteria but do not have SLE. These SLE «imitators» often baffle doctors, and patients may be misdiagnosis or even treated inappropriately.25
There are many similarities between the classification criteria for the diagnosis of SLE and TAFRO since both diseases present serositis, renal involvement, and thrombocytopenia.12,22 Fever, lymphadenopathies, and splenomegaly may also be common in patients with positive ANA who have SLE but may also occur in patients with TAFRO.23,26 Very few publications show the confusion that can occur to diagnose an extremely rare disease such as TAFRO in patients who meet qualifying criteria for SLE.27
Case presentation48-year-old man, journalist by profession from Popayan-Colombia. Otherwise, healthy and able to travel 20 kilometers by bike every other day up to one month before the first admission. He initially presented fever, cough, and nasal flow for three weeks that partially improved with the use of paracetamol and antihistamines then had lower limb edema, dyspnea, and progressive fatigue that was adding for six weeks from the initial symptoms appeared, that is why the patient was readmitted in emergency department on August 27, 2019.
The initial physical examination had hepatosplenomegaly, ascites, anasarca with bilateral pleural effusion, and peripheral edema. Main laboratory findings were anemia (hemoglobin 9.5g/dL), thrombocytopenia (28,000μL−1), CRP (5.71mg/dL), alkaline phosphatase alkaline (267U/L) and ferritin (1769ng/dL) elevated, with hypoalbuminemia (2.58g/dL) and severe renal dysfunction (creatinine 2.32mg/dL, blood urea nitrogen (BUN) 117.9mg/dL); without proteinuria, the GFR was 32.1mL/min/1.73m2, for severe renal dysfunction with anasarca, hemodialysis was initiated. HIV infection, hepatotropic viruses, and cytomegalovirus were ruled out. There was no isolation of fungi or bacteria in blood, urine, or ascitic liquid cultures (Table 1). Rapidly progressive glomerulonephritis was initially considered, so pulses of 1g/day of intravenous methylprednisolone (MTP) were administered three days. Low probability of thrombotic microangiopathy was considered, as the peripheral blood smear had no schistocytes, lactate dehydrogenase and bilirubin were normal, and direct coombs was weakly positive for c3d.
Laboratory findings.
| Variable | Result | Reference range |
|---|---|---|
| CBC | ||
| White blood cell count (cell/μL) | 8680 | 4230–10,070 |
| Hemoglobin (g/dL) | 9.5 | 13.7–17.5 |
| Hematocrit (%) | 29 | 40.1–51 |
| Platelets (cell/μL) | 28,000 | 163,000–337,000 |
| Neutrophils (cell/μL) | 5300 | 1780–5380 |
| Renal function | ||
| Serum creatinine (mg/dL) | 2.32 | 0.67–1.17 |
| BUN (mg/dL) | 117.9 | 6–20 |
| Urine protein (mg/24h) | 172mg/24h | 0–150 |
| Urinalysis | ||
| Density | 1.016 | 1.01–1.03 |
| PH | 5.0 | 4.5–8 |
| Nitrites | Negative | Negative |
| Proteins (mg/dL) | Negative | Negative |
| Glucose (mg/dL) | Negative | Negative |
| Bilirubin (mg/dL) | Negative | Negative |
| Hemoglobin (/μL) | Negative | Negative |
| Urine sediment | ||
| Leukocytes (XC) | 0–3 | 0–5 |
| Bacteria | + | Negative |
| Hyaline casts (XC) | >10 | Negative |
| Granular casts (XC) | 2–4 | Negative |
| Blood chemistry panel | ||
| CRP (mg/dL) | 5.71 | 0–0.5 |
| Alkaline phosphatase (U/L) | 267 | 40–129 |
| Gama glutamyl transferase (U/L) | 52 | 0–71 |
| Glutamic pyruvic transaminase (U/L) | 5 | 0–41 |
| Glutamic oxaloacetic transaminase (U/L) | 10 | 0–38 |
| Total bilirubin (mg/dL) | 0.38 | 0–1.2 |
| Direct bilirubin (mg/dL) | 0.28 | 0–0.3 |
| Lactate dehydrogenase (U/L) | 123 | 135–225 |
| Triglycerides (mg/dL) | 76 | 0–200 |
| Serology tests | ||
| Thyroid stimulating hormone (TSH) (μUI/mL) | 10.8 | 0.27–4.2 |
| Free thyroxine (T4F) (ng/dL) | 0.7 | 0.93–1.7 |
| Antithyroglobulin antibodies IgG (UI/mL) | 582.55 | <4.1 |
| Thyroperoxidase antibodies (UI/mL) | 25.31 | <5.61 |
| Direct coombs | C3d+ | Negative |
| Fibrinogen (mg/dL) | 256 | 200–300 |
| Ferritin (ng/mL) | 1769 | 30–400 |
| Rheumatoid factor (UI/mL) | 30 | <14 |
| ANAs (dilution, pattern) | 1:5120, speckled | Negative |
| DNA antibodies (UI/mL) | 2.2 | <20 |
| SSA(Ro) antibodies (UI/mL) | 3.5 | <15 |
| SSB(La) antibodies (UI/mL) | 5.9 | <15 |
| Sm antibodies (UI/mL) | 1.2 | <15 |
| RNP antibodies (UI/mL) | 112.5 | <15 |
| C1q antibodies (U/mL) | 8.4 | <10 |
| Myeloperoxidase antibodies (MPO) (U/mL) | 1.6 | <5 |
| Serine proteinase-3 antibodies (PR3) (U/mL) | 1.4 | <5 |
| Complement 3 (mg/dL) | 84.74 | 90–180 |
| Complement 4 (mg/dL) | 10.7 | 10–40 |
| Immunoglobin G (g/L) | 19.39 | 7–16 |
| Immunoglobin A (g/L) | 2.19 | 0.7–4 |
| Immunoglobin M (g/L) | 0.51 | 0.4–2.3 |
| Lupus anticoagulant ratio | 1.11 | <1.2 |
| Beta-2 Glycoprotein 1 antibodies IgG (U/mL) | 4.4 | <5 |
| Beta-2 Glycoprotein 1 antibodies IgA (U/mL) | 0.4 | <5 |
| Beta-2 Glycoprotein 1 antibodies IgM (U/mL) | 0.9 | <5 |
| Anticardiolipin antibodies IgG (GPL-U/mL) | 2.8 | <10 |
| Anticardiolipin antibodies IgG (MPL-U/mL) | 0.7 | <7 |
| Beta 2 microglobulin | 7.5 | 0.8–2.2 |
| Cryoglobulins | Negative | Negative |
| Smooth muscle antibody (dilution) | Negative | Negative |
| Anti-mitochondrial antibody (dilution) | Negative | Negative |
The patient had positive ANA with a dilution of 1:5120 speckled pattern, extractable nuclear antigen (ENA) antibody tests only had positive anti-ribonucleoprotein (RNP), also with C3 complement slightly below normal level and normal C4 complement levels. For positive ANAs, fever, pleural effusion, thrombocytopenia, and decreased C3 was diagnosed SLE, met 14 points of the classification criteria EULAR-ACR 2019.22 On suspicion of renal and hematological commitment for SLE and related secondary autoimmune thyroiditis, prednisone 50mg/day was continued after methylprednisolone pulses. The patient strangely did not have significant proteinuria in the nephrotic range, as is usually the case in patients with SLE-associated nephropathy. Thrombocytopenia and anemia were refractory to systemic corticosteroid treatment. Intravenous immunoglobulin G (IVIgG) 1g/kg/day was administered for two days, considering immune-mediated thrombocytopenia. To confirm lupus nephropathy's presence was performed renal biopsy after a platelet transfusion for renal hematoma risk. In renal histopathology, only acute tubular necrosis of 15% of the glomerulus evaluated was evident, and strikingly immunofluorescence was negative against SLE nephropathy diagnose (Fig. 1).

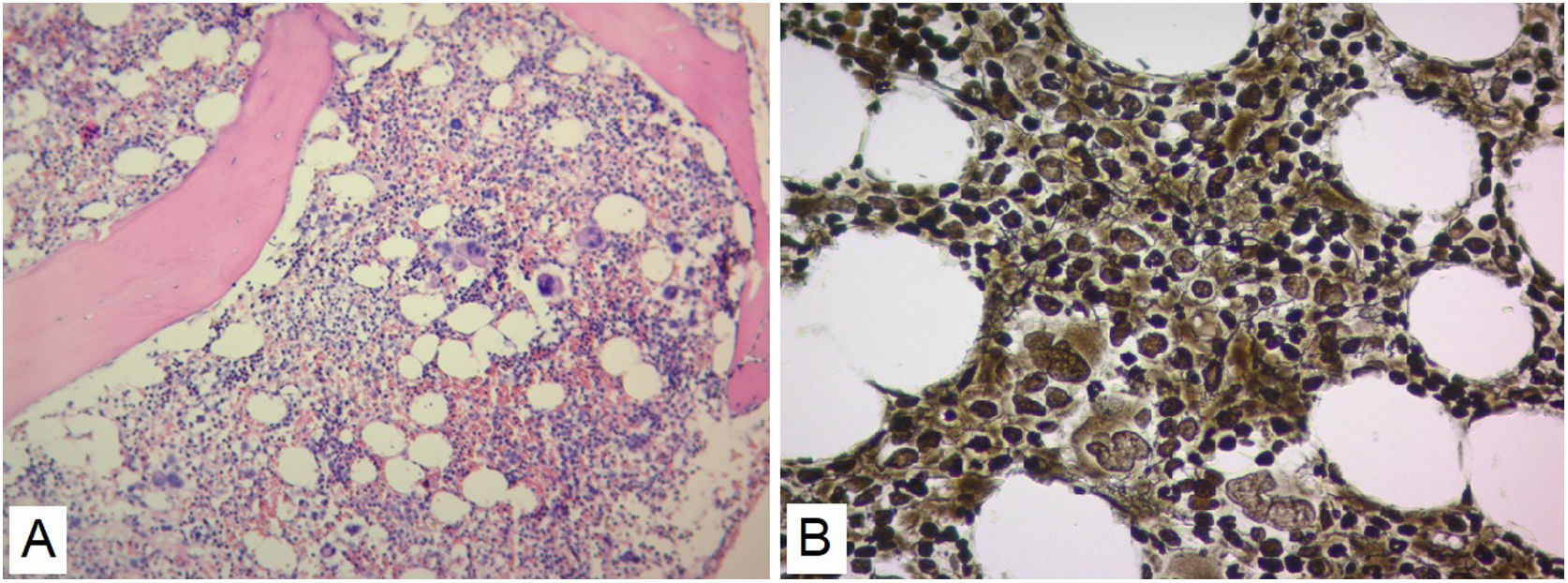
No evidence was found that the cause of renal failure was due to SLE or of infectious origin, and a secondary macrophage activation syndrome was refused as it did not find altered triglyceride and fibrinogen values (Table 1). The patient had insufficient improvement of thrombocytopenia and anemia with systemic corticosteroids and IVIgG, there was also no robust findings that the patient's bicytopenia was by hemolysis. The diagnostic possibility of a malignant hematological disease or other less common blood disorder was considered. A BMAB was performed, the flow cytometry was negative for hematological malignancy, and histopathological study showed hypercellular bone marrow with increased megakaryocytes, and reticulin fibers (Fig. 2). Flow cytometry studies for paroxysmal hemoglobinuria were negative.
.")
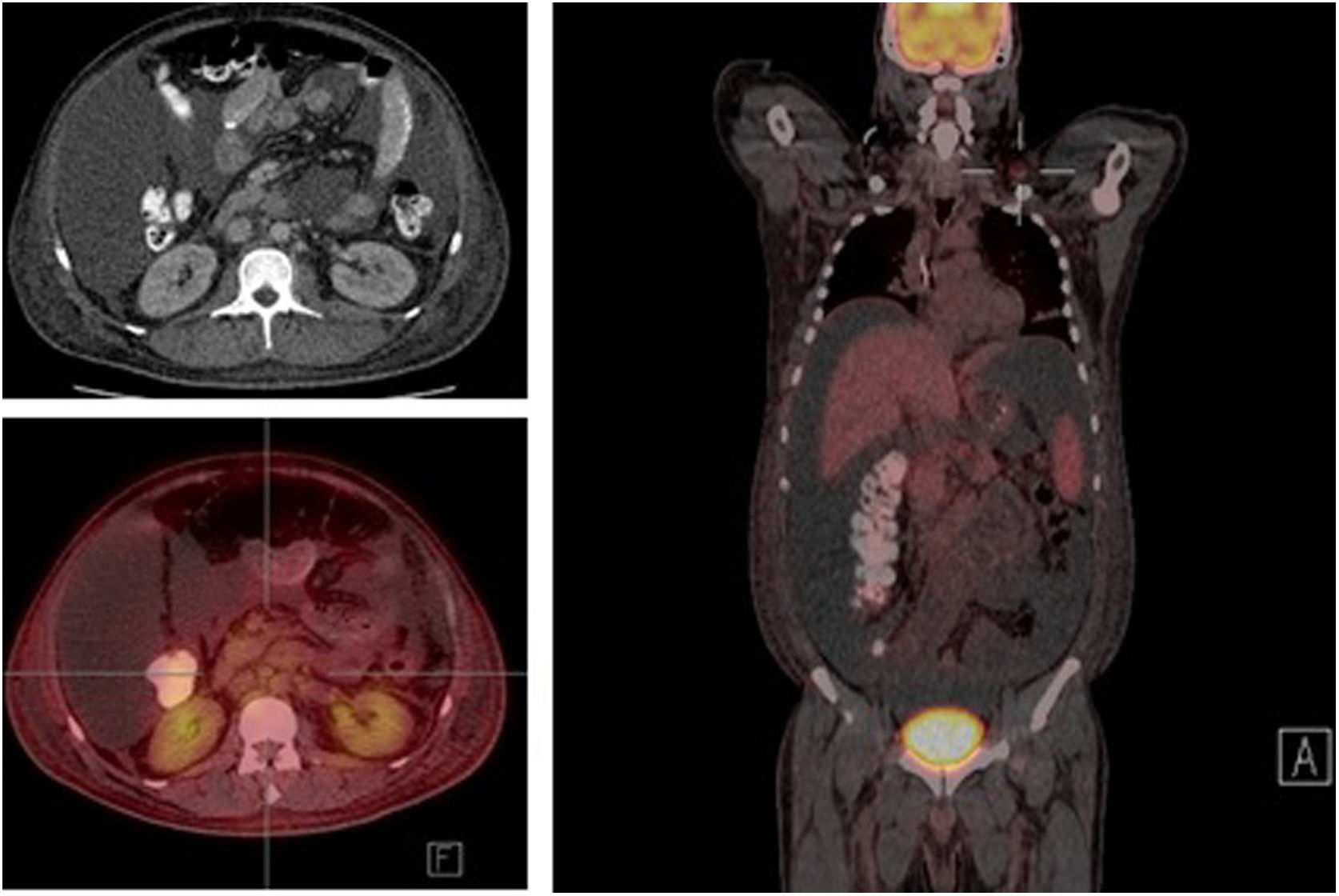
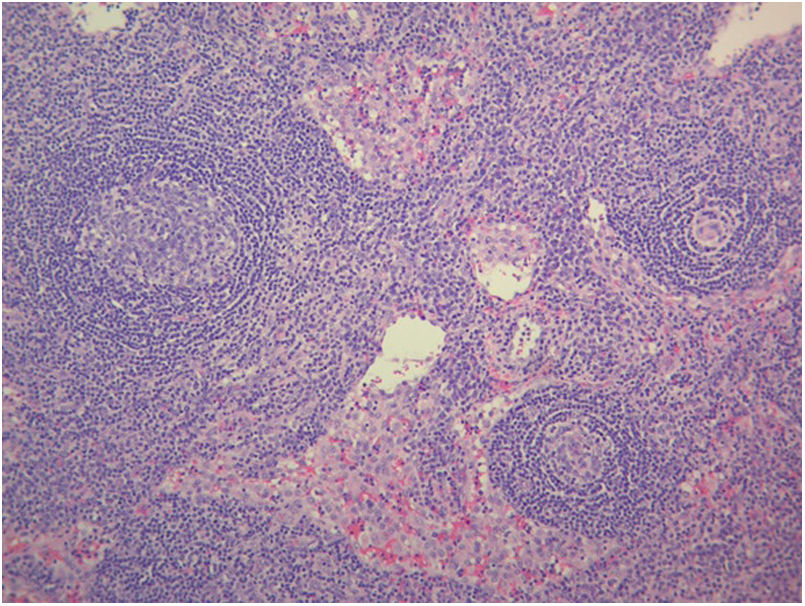
The study of solid tumor or hematological disease that was not represented in BMAB was performed with positron emission tomography (PET). In PET hypermetabolism of supra and infra diaphragmatic nodes with polyserositis was found (Fig. 3), excisional biopsy of the left supraclavicular node was taken, the histopathology showed changes suggesting Castleman disease hyaline-vascular variety, and latency-associated nuclear antigen (LANA-1) of HHV-8 was negative (Fig. 4).


The histopathology of the lymph node confirmed iMCD; the presence of hyaline-vascular variety associated with interfollicular vascular proliferation configured the typical lymph node biopsy of TAFRO. The patient fulfilled all diagnostic criteria for TAFRO, had histopathological criteria in lymph node and bone marrow, besides had thrombocytopenia less than 100,000μL−1, anasarca with pleural effusion and ascites documented in computerized tomography (CT) scan, increased fever of 38.0°C without clear origin, elevated CRP, progressive renal failure with hemodialysis necessity, besides organomegaly with splenomegaly and lymphadenopathy documented in CT scan. However, this patient also met the SLE classification criteria, an exclusion criterion for diagnosing TAFRO syndrome (Table 2).12 Furthermore, if the idea that the patient had a diagnosis of TAFRO syndrome was maintained, he presented anasarca with pitting edema, thrombocytopenia less than 50,000μL−1, CRP above 2mg/dL, and GFR less than 60mL/min/1.73m2, him would be classified as slightly severe TAFRO syndrome (Table 3).12 In a scenario where the patient met the criteria for both SLE and TAFRO, despite having severe kidney failure with polyserositis, it did not improve with pulse-dose steroids. Renal biopsy did not explain renal failure by possible diagnosis of SLE. The thrombocytopenia was considered immune-mediated; however, lactate dehydrogenase or bilirubin was not elevated and did not improve with systemic steroids or IVIgG. Although it had slightly decreased C3 with normal C4, it also had hypoalbuminemia, which decreased the likelihood of complement consumption by an active SLE. All evidence showed that despite the patient fulfilled the criteria to be classified as SLE, this was not the diagnosis and that the correct diagnosis was TAFRO syndrome. TAFRO might explain all the pathological findings in the patient and simulated the presence of SLE.
Diagnostic criteria for TAFRO syndrome.
| Diseases to be excluded |
| (1) Malignancies, including lymphoma, myeloma, mesothelioma, etc. |
| (2) Autoimmune disorders, including SLE, Sjögren's syndrome, ANCA-associated vasculitis, etc. |
| (3) Infectious disorders, including acid-fast bacterial infection, rickettsial disease, Lyme disease, severe fever with thrombocytopenia syndrome, etc. |
| (4) POEMS syndrome |
| (5) Hepatic cirrhosis |
| (6) Thrombotic thrombocytopenic purpura (TTP)/hemolytic uremic syndrome |
| Major categories: need all |
| (1) Anasarca, including pleural effusion, ascites, and general edema |
| (2) Thrombocytopenia; platelet count ≤100,000μL−1, without myelosuppressive treatment |
| (3) Systemic inflammation, defined as fever of unknown etiology above 37.5°C and/or serum C-reactive protein concentration ≥2mg/dL |
| Minor criteria: at least 2 of 4 |
| (1) Castleman disease-like features on lymph node biopsy |
| (2) Reticulin myelofibrosis and/or increased number of megakaryocytes in bone marrow |
| (3) Mild organomegaly, including hepatomegaly, splenomegaly, and lymphadenopathy |
| (4) Progressive renal insufficiency |
Modified from Masaki et al.12
Disease severity classification for TAFRO syndrome.
| (1) Anasarca: three points maximum | ||
| One point for pleural effusion on imaging | ||
| One point for ascites on imaging | ||
| One point for pitting edema on physical examination | ||
| (2) Thrombocytopenia: three points maximum | ||
| One point for lowest platelet counts <100,000μL−1 | ||
| Two points for lowest platelet counts <50,000μL−1 | ||
| Three points for lowest platelet counts <10,000μL−1 | ||
| (3) Fever and/or inflammation: three points maximum | ||
| One point for fever ≥37.5°C but <38.0°C or for CRP ≥2mg/dL but <10mg/dL | ||
| Two points for fever ≥38.0°C but <39.0°C or for CRP ≥10mg/dL but <20mg/dL | ||
| Three points for fever ≥39.0°C or for CRP ≥20mg/dL | ||
| (4) Renal insufficiency: three points maximum | ||
| One point for GFR <60mL/min/1.73m2 | ||
| Two point for GFR <30mL/min/1.73m2 | ||
| Three points for GFR <15mL/min/1.73m2 or in need of hemodialysis | ||
| Relationship between score and disease severity | ||
| 0–4 points | Mild | Grade 1 |
| 5–6 points | Moderate | Grade 2 |
| 7–8 points | Slightly severe | Grade 3 |
| 9–10 points | Severe | Grade 4 |
| 11–12 points | Very severe | Grade 5 |
Modified from Masaki et al.12
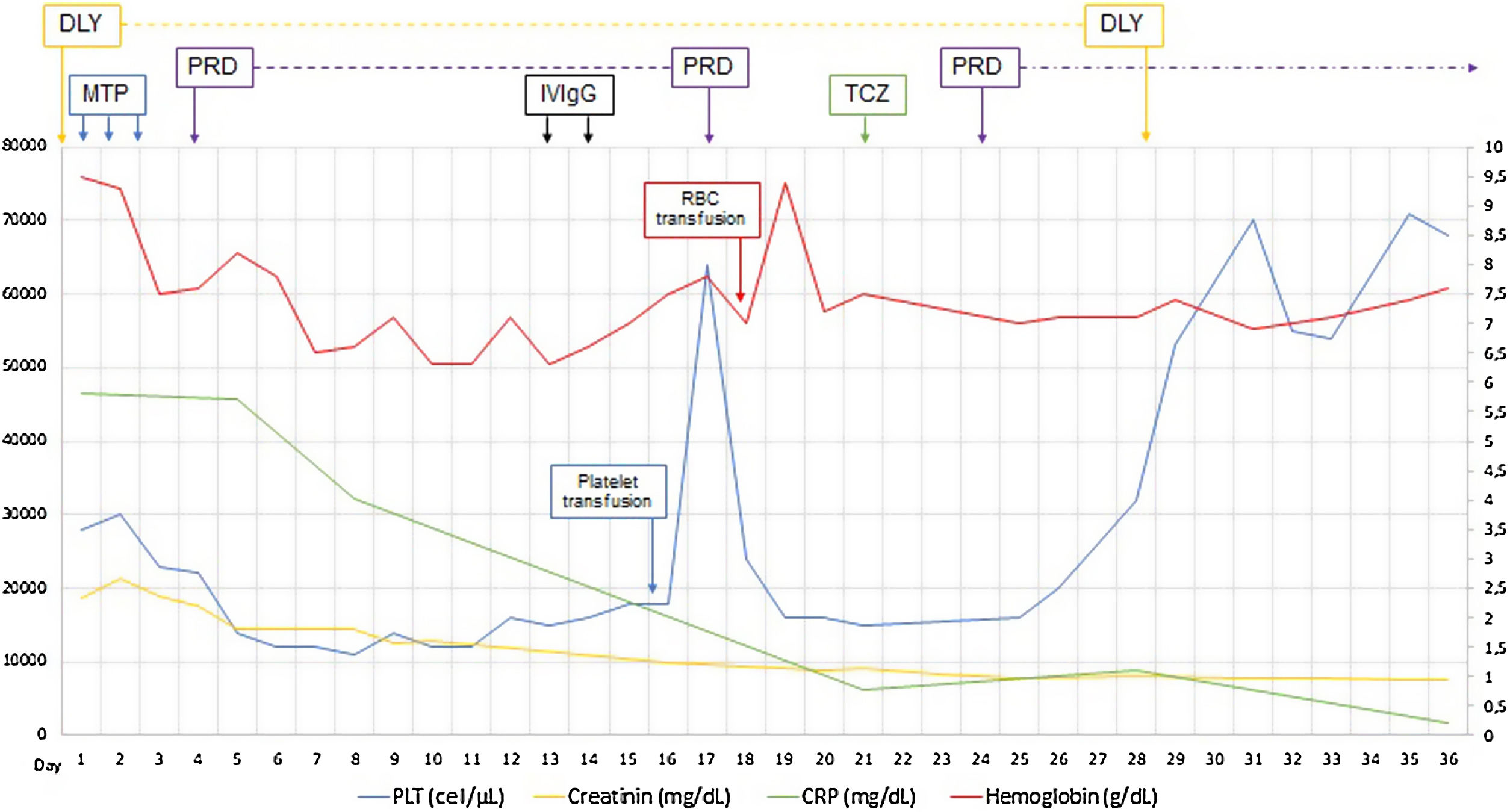
Established the diagnosis of TAFRO syndrome and tocilizumab was started, defined as slightly severe and in the absence of treatment guidelines the usual dose of 4mg/kg every four weeks was chosen, and because persisted the low amount of platelets was restarted three days later oral prednisone 1mg/kg/day. After initiating the treatment, the patient improved platelet and hemoglobin levels, decreased serum creatinine and CRP, discontinued hemodialysis, and was discharged at day 36 of admission (Fig. 1). The patient continued treatment on an outpatient basis but died three months after being discharged by intracerebral hemorrhage.
DiscussionWe present the case of a 48-year-old man with TAFRO syndrome who also met the diagnostic criteria of SLE. This case highlights important clinical knowledge, such as that TAFRO syndrome can simulate SLE and should be taken into account as differential diagnosis, since treatment requires an entirely different approach, and the typical TAFRO syndrome patient is not the same as in SLE.
TAFRO syndrome is an extremely rare disease. The incidence of Castleman disease in the United States is 21.7–25.9/1 million inhabitants, of which only 23%; between 4.2 and 5.4/1 million inhabitants correspond to MCD, within the MCD group, iMCD accounts for approximately 30% of cases with 1.26–1.62/1 million inhabitants, and in the latter group some studies estimate that about 2% of cases iMCD correspond to TAFRO syndrome.19 It can then be calculated that the global incidence of TAFRO would be 197 to 253 cases for a global population estimated by 2020 of 7.8 billion.28 There are only approximately 100 cases of TAFRO reported throughout the world medical literature. The publication with the highest number of patients is a retrospective study conducted in Japan by Fujimoto et al. with 82 patients.17,29 In Latin America only two cases of TAFRO syndrome are reported, both reported in 2017, the first reported at the Albert Einstein hospital in Brazil by Freire et al.,30 and the second reported at the Fundación Oftalmológica de Santander Carlos Ardila Lϋlle in Colombia by Ortiz et al.31 This is the third case of TAFRO syndrome reported in Latin America and the first report that meets the SLE classification criteria.
The IL-6 importance has been widely documented in Castleman disease, where HHV-8 stimulates the production of a form of IL-6 encoded in the viral genome. The viral IL-6 and other stimuli such as vascular endothelial growth factor (VEGF) generate the production of IL-6 in large quantities in patients with Castleman disease. The cases of MCD are usually related to high serum IL-6 and VEGF levels. However, in cases of iMCD, the disease is conducted by different mechanisms such as neoplastic, autoimmune, or by virus infection different than HHV-8; that mechanism stimulates the production of IL-6 and VEGF, producing the same clinical characteristics as MCD,18,32 and that is why the treatment is based on blocking IL-6 with siltuximab or its receptor with tocilizumab.26 In TAFRO syndrome, it is known that IL-6 explains a large part of the findings in all MCD, but in TAFRO, there are not always high levels of IL-6, which suggests that other pro-inflammatory cytokines such as IL-2 and tumor necrosis factor-α (TNF-α) have a role in the development of the disease and the expansion of the inflammatory response.10,13 A recent study by Langan et al. showed that the induction of the mTOR pathway by type I interferon has an essential relationship with the development and severity of manifestations in TAFRO,15 which could better explain the significant variability in the clinical presentation and severity of this syndrome.
Several cases of TAFRO syndrome simulating other diseases are reported. Concerning SLE, the relationship is more complicated since TAFRO syndrome is usually described as a disease with some manifestations that can be confused with autoimmune disease,33,34 but it is clear that to diagnose TAFRO syndrome is necessary to exclude SLE (Table 2). A case of a 48-year-old male patient who met the diagnostic criteria for SLE and had all the findings of TAFRO syndrome is presented. Cases like this are exceptional and represent a challenge for rheumatologists since they should define the cause of the patient's clinical condition with precision and giving treatment in consequence. Hasegawa et al. conducted a retrospective study with 46 patients diagnosed with SLE; 4 met TAFRO criteria. These four patients were men over 50 years old with a new diagnostic of SLE.27 Rheumatologists should be mindful of TAFRO syndrome diagnosis in patients with unusual SLE presentations that do not improve with usual treatment, especially if they are middle-aged men with recent manifestations.
A possible confusion between TAFRO and SLE is in the BMAB. In the histopathological study of bone marrow is evaluated the level of fibrosis and reticulin fibers’ presence, this scaffolding based on collagen type I and III is an indirect view of cellular bone marrow microenvironment. The bone marrow hypercellularity given by any cause generates changes that will require more significant support of reticulin and collagen, which changes over time tend to be permanent generating fibrosis. On the one hand, the proportions of reticulin and collagen, and on the other hand, the level of fibrosis, are two markers that differentiate between healthy and sick patients. Many causes can generate changes in the bone marrow's microenvironment, such as hematological neoplasms or preneoplastic hematological conditions. However, virtually any situation that causes chronic inflammation can produce reticulin fibrosis changes, or even myelofibrosis, autoimmune diseases such as SLE, systemic sclerosis, Sjögren's, or antiphospholipid syndrome. Also, calcium metabolic disorders such as hyperparathyroidism or vitamin D deficiency, or granulomatous infections such as tuberculosis can be counted. Bone marrow stromal fiber disorders are usually associated with abnormalities in megakaryocyte and platelets’ number or function, and cytokines from that cells appear to be necessary but not enough to occur fibrosis. In situations such as TAFRO in which thrombocytopenia occurs, platelet-derived growth factor contained in megakaryocytes and platelets is released, which among other functions, is a potent stimulator of fibroblast growth; hence, this cytokine has been proposed as a possible cause of bone marrow fibrosis. Furthermore, transforming growth factor-β (TGF-β) is a protein with a latent activity that is activated through acidic environments or proteases. TGF-β is also found in megakaryocytes and platelets, and when is activated, it is a potent stimulator of fibroblasts synthesis of collagen; this has been demonstrated in vitro with the stimulation of bone marrow fibroblasts by TGF-β, where more type III procollagen and reticulin are produced than type I procollagen and collagen.35,36
The histopathological findings in lymph nodes are different in SLE respect of TAFRO syndrome. SLE lymphadenopathy occurs in approximately 60% of patients, where reactive nodal hyperplasia is common, but this is not a specific finding. The most specific SLE finding is coagulation necrosis but is not frequently observed, histologically has been described varying degrees of coagulation necrosis with hematoxylin bodies in lesions of lymph nodes, which is a finding being exclusive to SLE.37 TAFRO syndrome is defined with a typical form of lymph node, presenting a considerable increase of interfollicular vessels of the upper endothelium's venules with germinal center reaction, but with atrophy of that germinal centers, which differentiates it from iMCD-NOS.10,33
The germinal center reaction is evidenced in both SLE and TAFRO syndrome, in TAFRO could explain a large part of its clinical manifestations. IL-6 has an essential role in developing the germinal center; it stimulates B-Cell Lymphoma 6 (BCL-6) expression in CD4+T lymphocytes, polarizing this lineage toward follicular T lymphocytes, thus promoting the formation of the germinal center.38 In normal conditions, interferon-γ (IFN-γ) synergizes with signals from the B cell receptor (BCR), toll-like receptor (TLR), and CD40 to promote the production of IL-6 from B lymphocytes and thus contribute to the formation of the germinal center, SLE patients that have high levels of IFN-γ may present more aggressive forms of the disease, although SLE is multifactorial, cytokines such as B cell activator factor (BAFF), IL-6, IL-17, IL-21, IL-8, IFN-γ, and endosomal TLR 7 and 9 are an important part of the development of the disease.39,40 In SLE, B lymphocytes are the primary source of IL-6, which is essential for developing spontaneous autoreactive germinal centers and inducing the recombination activating gene (RAG), stimulating the production of autoantibodies. In murine models of SLE, blocking IL-6 derived from B lymphocytes prevents glomerulonephritis due to immune complex deposits.39 In TAFRO syndrome, IL-6 is one of the pillars of its pathophysiology. There are changes similar to those of SLE that can cause lymphadenopathy, although in TAFRO, it is likely that the expression of BAFF, CXCL13, or IFN-γ does not occur, which would explain the constant appearance of germinal centers that cannot be maintained in the time. The germinal center reaction beginning without achieving its maintenance may be the reason for the finding of atrophic germinal centers, that could also explain the normal or slightly elevated levels of serum gamma globulins in TAFRO as a complete germinal center reaction cannot be generated, besides the inconstant or absent stimulus for the isotype immunoglobulins change. The theory of the incomplete germinal center formation could also explain in part the presence of high ANA titers in this patient, since when cytokines such as BAFF are not expressed, massive apoptosis of plasmablasts occurs, which can be a source of autoantigens that would generate a positive feedback to IL-6 initiated B cell-dependent reaction. The presence of high titers of ANA has not been previously documented and is one of the points that most can generate confusion between diagnoses of TAFRO syndrome and SLE. The incomplete germinal center reaction provides insight into understanding this overlap between the diseases.
Organomegaly is not usual in SLE, but it is one of the defining characteristics of TAFRO. The organomegaly is explained by the excessive production of IL-6 and VEGF that can alter systemic lymphatic and vascular permeability, generating anasarca. Besides, changes in lymphatic and vascular permeability also cause edema within solid organs, producing organomegaly, these changes generate hydrodynamic alterations like those that occur in cirrhotic patients, but in the absence of liver damage. The generalized alteration in vascular permeability explains findings such as the ascites and pleural effusion, which in the context of a patient with hyperproduction of pro-inflammatory cytokines can produce secondary serositis that can be confused with the findings of SLE. However, the serositis in SLE has a different cause explained by deposits of immune complex in these areas. On the other hand, renal failure in TAFRO is not associated with the deposit of immune complex or glomerular inflammation as in SLE, renal failure in TAFRO is more similar to hepatorenal syndrome, where changes in vascular permeability decreased intravascular volume, besides the possible direct compression on the renal vessels by lymph node conglomerates, it is for these reasons that acute tubular necrosis may be a common finding in patients with renal failure due to TAFRO.40–42
Thrombocytopenia in TAFRO can be complicated to explain since, in iMCD-NOS, thrombocytosis is usual. Thrombopoiesis is influenced by multiple cytokines such as IL-3, IL-6, IL-11, granulocyte-monocyte colony-stimulating factor, erythropoietin, and thrombopoietin. Moreover, although the IL-6 intervenes in all phases of megakaryocytopoiesis, there are counter-regulatory mechanisms based mainly on thrombopoietin levels that work by different pathways.43 The direct stimulus related to IL-6 levels may produce thrombocytosis in iMCD-NOS, but in TAFRO syndrome could stimulate a consumption thrombocytopenia related to an autoimmune phenomenon induced by the serum cytokine environment. Besides, although TAFRO patients have megakaryocytic hyperplasia in bone marrow, they also have a low platelet-derived growth factor level; thus, increased consumption is not the only cause of thrombocytopenia, and megakaryocytes dysfunction also can contribute to that situation. That dysfunction could explain the inadequate response to thrombopoietin receptor stimulators and platelet transfusions,1,17,36 which could mean an additional difference between iMCD-NOS and TAFRO.
The first line of treatment for TAFRO syndrome is systemic glucocorticoids with a 40–60% success rate.13,20,44 Immunosuppressive regimens have been used for cases that do not respond to steroids. Treatments have reported in literature include ciclosporin,3,20,45 sirolimus,15,26,46 anakinra,26 bortezomib,47 anti-IL-6 monoclonal antibodies (siltuximab), anti-IL-6 receptors (tocilizumab),5,48,49 and anti-CD20 (rituximab) with successful results in case reports.50 However, combined regimens with thalidomide51 or cytotoxic agents such as cyclophosphamide52,53 also have been reported for refractory cases. In very severe cases (Table 3) have been employed plasma exchange54 associated with immunosuppressive treatment or critic care therapy such as extracorporeal veno-arterial membranous oxygenation.55 Some publications suggest bone marrow transplantation or chemotherapy use, recommending R-CHOP (Rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) or R-CVP (rituximab, cyclophosphamide, vincristine, prednisolone).14,56 In 2019 Matsuhisa collected data from 23 published cases that met the diagnostic criteria for TAFRO syndrome and described the treatment response. Platelets count increased >100,000μL−1, and anasarca resolved in all survivors; however, this happened several weeks after started the treatment (between 3 and 20 weeks). Nevertheless, care should be taken with excess immunosuppression, and as when CRP levels decrease to <1.0mg/dL immediately after the initiation of treatment, it can be an early indicator of mortality associated with excess immunosuppression.53
TAFRO syndrome treatment is based on report cases because treatment guides or consensus do not exist. The most important is to identify severe and non-severe cases (Table 3), and the goal is to achieve complete remission. Complete remission is defined as recovery of platelet count, hemoglobin level, edema, organomegaly, renal failure, besides fever remission, and CRP decrease.13,14 In non-severe cases, the use of systemic steroids with prednisone doses of 1mg/kg/day or its equivalent in addition to siltuximab or tocilizumab is recommended in the first line due to the important role of IL-6 in the physiopathology of TAFRO. Also, it is possible to use the proposed dose of systemic steroids in addition to a short cycle of rituximab 375mg/m2 dose each week for 4 to 8 weeks. Rituximab should not be used for severe cases because of its delay in the onset of its therapeutic effect.14,50 In severe cases, it should be initiated with the systemic steroid scheme associated with siltuximab or tocilizumab in first line. Standard tocilizumab dose is no published, but report-based management and our own bad experience with this case suggest a dose of 8mg/kg every 2–4 weeks, and in case of no complete remission, the use of a mixture of immunomodulators have been documented.14,34 Whether complete remission is achieved, the patient should be followed weekly, and it is also possible to reduce the dose of tocilizumab up to 4mg/kg/every four weeks and make a progressive tapering of steroids according to clinical response, platelet count, hemoglobin level, and CRP levels.34
ConclusionTAFRO syndrome is an exceedingly rare disease that can be confused with SLE. TAFRO syndrome should be considered in patients with unusual SLE manifestations that do not agree with the severity of the case, especially in men around 50 years or older diagnosed with SLE de novo. The presence of ANA in TAFRO syndrome has not been documented, and this case shows that it is possible, a TAFRO syndrome diagnosis that also meet SLE classification criteria as pathophysiological mechanisms may overlap. However, the treatment has a different approach, and mortality in TAFRO syndrome is high in severe cases that are not adequately treated in time. This case invites to reconsider the diagnostic criteria for TAFRO in which SLE should be ruled out.
FundingClinical case considered the best in the national rheumatology residents’ competition 2020. Awarded thanks to the funding of the Colombian Rheumatology Association (ASOREUMA).
Consent for publicationWritten informed consent was obtained from the patient for publication of this case report and any accompanying images.
Conflict of interestsThe authors have no conflict of interest to disclose.
To Fundacion Valle del Lili Universitarian Hospital, and ASOREUMA for supporting high-level medical education in rheumatology.
