



Optic neuromyelitis (ONM), also called neuromyelitis optica spectrum (Neuromyelitis Optica Spectrum Disorders, NMOSD) is recognized as an inflammatory autoimmune demyelinating disease of the central nervous system, mediated by autoantibodies against the aquaporin-4 receptor (AQP4-IgG). It predominantly affects the optic nerves and the spinal cord.1–3 It is known that patients with immune disorders are more likely to present other autoimmune diseases, but the relation between juvenile idiopathic arthritis and ONM has not been completely described.5 In this paper, we report a case of a patient with juvenile idiopathic arthritis, presenting with a rapidly progressive neurological condition, who is treated with biological drugs.1–4
La neuromielitis óptica (NMO), también llamada espectro de la neuromielitis óptica (neuromyelitis optica spectrum disorders, NMOSD) se reconoce como una enfermedad inflamatoria, autoinmune, desmielinizante del sistema nervioso central, mediada por autoanticuerpos contra el receptor de acuaporina 4 (AQP4-IgG) que afecta predominantemente a los nervios ópticos y la médula espinal1–3. Es conocido que los pacientes con trastornos inmunitarios, tienen más probabilidades de presentar otras enfermedades autoinmunes, sin embargo, no está completamente descrita la asociación entre artritis idiopática juvenil y NMO5. En este escrito se reporta el caso de una paciente que cursa con artritis idiopática juvenil, debuta con compromiso neurológico rápidamente progresivo, y es tratada con medicamentos biológicos1–4.
Neuromyelitis optica (NMO), initially known as Devic’s disease or Devic’s syndrome, is an autoimmune inflammatory demyelinating disease of the central nervous system (CNS) that predominantly affects the optic nerves and the spinal cord. Although Thomas Clifford Allbutt described the association between unilateral optic nerve disorder and myelitis in 1870, it was the French scientist Eugene Devic who used the term “neuromyelitis optica” for the first time, which was classified initially and until less than a decade ago as a variant of multiple sclerosis (MS) with involvement of the optical nerve4; however, today it is known that it is a different entity.5
In 2015, the new diagnostic criteria for this disease were published,6 which highlight the importance of the positivity of the serum autoantibodies targeted to the aquaporin-4 (AQP4-IgG) channel, accompanied by the clinical manifestations and the lesions observed in magnetic resonance imaging (MRI), for early diagnosis, which constitutes a determining factor in the prognosis of the patient.7–10 The comorbidity of NMO with other autoimmune diseases has been considered a factor for poor prognosis; however, the presence of AQP4-IgG alone does not support this association. It has been related to different autoimmune processes, such as systemic lupus erythematosus (SLE), Sjögren's syndrome (SS), myasthenia gravis (MG) and vasculitis, among others. Its association with rheumatoid arthritis (RA) has been described more recently.11
The purpose of this paper is to document a case of NMO in a patient with a history of juvenile idiopathic arthritis previously treated with multiple biological drugs.1,12–14
Presentation of the clinical caseA 45-year-old female patient with a previous diagnosis of juvenile idiopathic arthritis of 30 years of evolution, treated with chloroquine, azathioprine and prednisolone. She had received previously methotrexate, prednisolone, etanercept, adalimumab, leflunomide, and tocilizumab, with which no improvement in symptoms was observed, and also described adverse effects such as skin rash, drowsiness, loss of appetite, and alopecia.
The patient was admitted to the Emergency Department due to a worsening of the clinical picture that had started 10 months earlier with the presence of paresis of the right lower limb. Currently, she consulted due to paraparesis and loss of sphincter control with a T8 sensory level. The diagnosis of longitudinally extensive myelitis was established due to involvement of more than 3 vertebral segments, evidenced in the MRI, for which hospitalization was indicated in order to perform extension studies and start methylprednisolone 1 g IV every 24 h for 5 days.
A lumbar puncture was performed, which yielded results within normal limits, highlighting the absence of oligoclonal bands in the cerebrospinal fluid. Likewise, an electromyography of the 4 extremities was carried out with the incidental finding of involvement of sensitive median nerves with a myelin pattern. In addition, antinuclear antibodies (ANA), anti-DNA antibodies and complement, thyroid profile and levels of vitamin B12 were reported within normal parameters (Table 1).
Relevant paraclinical tests performed to the patient.
| Paraclinical test | Result |
|---|---|
| CRP | 96 |
| ESR | 38 mm/h |
| Rheumatoid factor | 120 IU/mL |
| Serum iron | 81 µg/dl (normal) |
| Vitamin B12 levels | 459 pg/dl |
| C3 | 115 mg/dl |
| C4 | 20.6 mg/dl |
| Total proteins | 4.5 g/dl |
| Albumin | 3 mg/dl |
| Globulin | 1.5 |
| A/G ratio | 2 |
| Free T3 | 3.8 (normal) |
| Free T4 | 0.97 ng/dl (normal) |
| TSH | 1.0 µIU/mL (normal) |
| Anti-DNA | Negative |
| ANA | Negative |
| Anti-Epstein-Barr virus antibody | Negative |
| AgsHb | Negative |
| Antibody against hepatitis C | Negative |
| Anti-aquaporin 4 antibodies | 59.4 (positive) |
| Ionic calcium | 1.3 mmol/l (normal) |
| Inorganic phosphorus | 1.8 mg/dl (low) |
| Protein electrophoresis in CSF | Did not show oligoclonal bands |
| Random urine proteins | 60 mg/dl (increased) |
| Anti-RNP antibodies | 7.2 IU (negative) |
| Anti-Ro antibodies | 9.1 IU (negative) |
| VDRL | Non-reactive |
| HIV | Negative |
| Bacilloscopy | Negative |
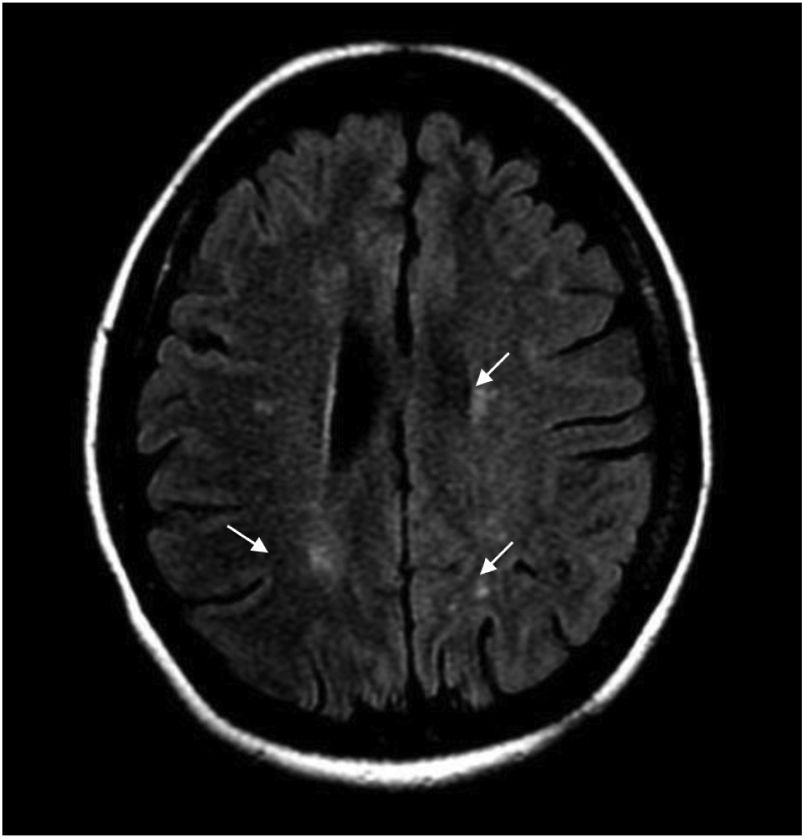
During hospitalization, the patient evolved torpidly, presented with neuropathic pain and the sensory level worsened to T6. Contrast-enhanced brain MRI was performed, finding multiple focal paraventricular and corticosubcortical images with hyperintensity in FLAIR sequence, without enhancement after administration of IV contrast medium or restriction in diffusion sequences (Fig. 1).
.")
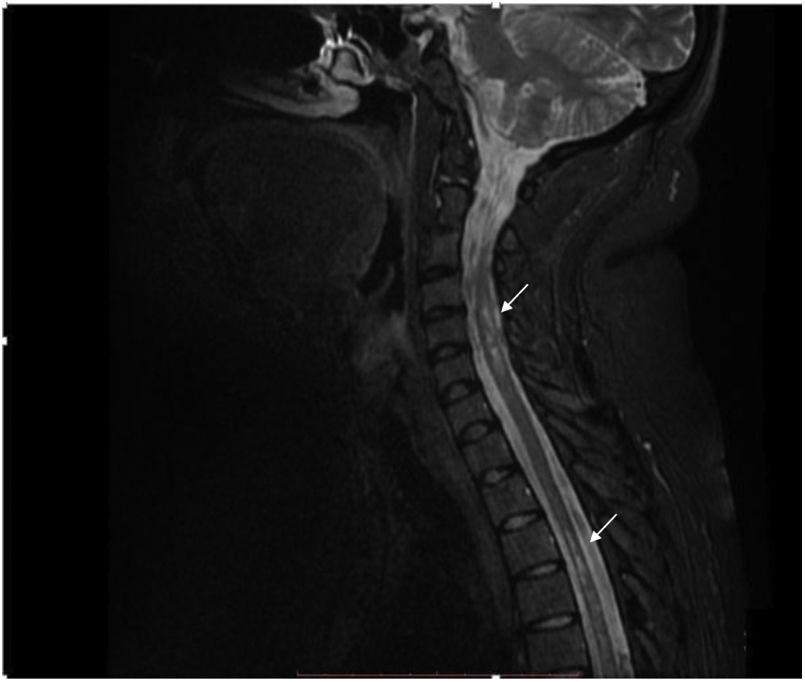
The contrast-enhanced spinal MRI showed hyperintense focal images at the level of the spinal cord, located predominantly laterally and diffusely distributed in the cervical and thoracic spine on the T2 sequence (Fig. 2). The diagnosis of longitudinally extensive transverse myelitis in a patient with a history of juvenile idiopathic arthritis was considered.
.")
The patient was assessed by the services of Internal Medicine, Rheumatology and Neurology, and the latter requested serum levels of AQP4-IgG, that were elevated (59.4 U/mL), which allowed to establish the diagnosis of NMO. Therefore, it was decided to held a medical board for the establishment of the treatment of the patient, and Rheumatology and Neurology agreed to start biological therapy with rituximab, due to the history of use of multiple biological and anti-TNF agents without obtaining a good response, since the patient, apart from the neurological manifestations already described, presented ulnar deviation of the metacarpophalangeal joints, swan-neck deformity and intense pain in the aforementioned joints.
Management with rituximab was started as follows: 2 doses of 1 g, separated by an interval of 15 days, and subsequently 1 g every 6 months. During the one-year follow-up, no new relapses were observed, however, the patient was left with significant sequelae that made her completely dependent for activities of daily living.
DiscussionThe autoimmune origin of NMO has been recognized; it is a fairly complex demyelinating and inflammatory disease, which presents an interaction between genetic and environmental factors.10 It follows a relapsing course in more than 80%–90% of cases, but its incidence is not clearly established due to misdiagnosis as MS. It is more prevalent in non-Caucasians and in females, with a 9:1 ratio in this second case. The age of onset ranges from childhood to adulthood and primarily affects young adults, with a mean age in the United States of 41.1 years, in contrast with the age of onset of MS which is generally 10 years before.1,7,14
Several studies show an incidence that varies from 0.053 to 0.4 per 100,000 people and prevalence rates that range between 0.52 and 4.4 per 100,000 people, which makes it an orphan disease.13 However, the search for a diagnosis with a higher degree of certainty in the shortest time possible has led experts to develop the new criteria for NMO.
Prior to the 2015 consensus, NMO was considered to be a disease with a monophasic course that required involvement of the optic nerve and the spinal cord; however, with the subsequent descriptions, new MRI findings were found that revealed involvement of the CNS, which may be more restricted or more extensive than that demonstrated in the optic nerve and the spinal cord. Along with these imaging findings, there was the discovery of detectable serum antibodies that are targeted to the AQP4 channel, which are positive in the majority of patients with NMO.
Thus, in 2007 the term “neuromyelitis optica spectrum disorders” (NMOSD) was introduced, with which it was sought to include those patients seropositive for AQP4-IgG, with limited or inaugural forms of NMO, who were at high risk of future attacks. Patients with atypical NMO lesions (cerebral, diencephalic and in the brainstem) and those with simultaneous autoimmune disorders, as is the case of the reported patient, were also included, without disregarding those who presented opticospinal MS. With the criteria of 2015, the terms NMO and NMOSD were unified, given the diagnostic uncertainty and the possible heterogeneity of seronegative NMOSD, thus dividing the spectrum of the disease into those with NMOSD with AQP4-IgG and those with NMOSD without AQP4-IgG, which allowed that only one compatible clinical characteristic was required to make the diagnosis.6,15
In the case reported here, the diagnostic suspicion was confirmed by associating the clinical manifestations of longitudinally extensive transverse myelitis, high titers of the specific antibody, and the characteristic brain lesions previously described. It should be noted that the diagnosis of this type of diseases continues to be a challenge; however, the presence of the AQP4-IgG allowed us to confirm the diagnosis, bearing in mind that these are found in up to 75% of patients with NMOSD.3,16,17
In Colombia there are characterization data with the new criteria, according to the 2015 International Consensus, which report findings similar to those of other populations, both in the clinical and in the laboratory and imaging data. A study that evaluated a cohort of 22 patients, mostly women (86%), with an average age of onset of the disease of 31 years, was published in 2016. It was found that none of these patients had any comorbidity, although some of them had positive anti-DNA, ANCA, anti-Ro and ANA values; therefore, the possibility of association with other autoimmune processes was not ruled out.18
Here we report a case that confirms the possibility that these 2 autoimmune pathologies occur associated. It should be noted that even though it is recognized that a positive ANA result is not pathognomonic for a particular disease, it can be useful and should be interpreted in the context of the clinical presentation. Low titers or concentrations of ANAs can also be found in “normal” individuals, sometimes transiently, especially in women over 65 years of age.19
In the current literature, is corroborated that there is a strong association between NMO and other systemic autoimmune diseases; however, the data directly related with juvenile idiopathic arthritis and NMO are limited, which distinguishes the presentation of our patient, who also had a rapidly progressive course.11,14
To date, various autoimmune diseases have been reported in approximately 30% of the patients with NMO. Among the main ones are SLE, SS, MG, antiphospholipid syndrome, ANCA-associated diseases, Hashimoto's thyroiditis, pernicious anemia, idiopathic thrombocytopenic purpura, primary sclerosing cholangitis, and sarcoidosis. The foregoing suggests a genetic predisposition to polyautoimmunity, but further studies are needed to demonstrate and validate the association between NMO and RA, although today it is known that in both entities there is an alteration of humoral immunity.8,13,20–23
The importance of B-cells mediated humoral immunity in the pathogenesis of NMO has been described recently; therefore, once the diagnosis of NMO is established, immunosuppressive therapy should be started as soon as possible, seeking to delay the time of relapse, reduce the severity of future recurrences, and minimize permanent disability. There are several immunosuppressive agents that have been used in the treatment of NMO, such as rituximab, mycophenolate, azathioprine and mitoxantrone25. Among these, rituximab is the therapeutic option of choice in patients with autoimmune diseases, since it is a safe and effective therapy both in RA and in NMO; in Colombia, it has been widely used with good results.18 It is recognized for being the first mouse-human chimeric monoclonal antibody specific for the CD20 antigen on B lymphocytes that exerts control of these through the depletion of B cells by cytotoxicity.
It has been demonstrated that rituximab reduces the frequency and severity of the relapses in patients with NMO. On the other hand, this anti-CD20 produces clinical improvement in patients with RA,25 which is supported by various clinical trials since 1998, the year in which the efficacy of rituximab in RA was reported for the first time, recognizing its effectiveness in patients in whom TNF inhibitors have failed.24 In this way, it becomes the most appropriate medication for our patient, who had presented secondary reactions to other first-line medications, and it was also necessary to reduce the possibility of relapse, since the sequel disability was severe in her case. It is known that in patients with NMOSD, rituximab had demonstrated a marked and sustained reduction in the annual relapse rate, independently of the induction and maintenance regimens.25
ConclusionsThe strong association between NMO and other systemic autoimmune diseases has already been described, however, its concomitance with RA is still poorly understood, which is why it is important to continue describing case reports such as the one presented, in favor of a better understanding of possible risk factors, pathophysiology and treatment in this type of diseases, in which polyautoimmunity is a characteristic. The efficacy and safety shown by the advent of biological therapies in these cases, specifically rituximab, with an indication for both RA and NMO, are highlighted.
Ethical ConsiderationsThe patient authorized the use of her clinical history for publication.
Conflict of interestThe authors declare that they have no conflict of interest for the preparation of this paper.
