



Myositis ossificans is a benign disorder characterised by the formation of heterotopic bone in skeletal muscle or soft tissues. It is extremely rare in children, <1% of cases occur in children under 10 years. We present a 17-day-old boy that, after 10 days of Intermediate Care Unit stay, was referred to our hospital for a developmental dysplasia of the hip. On clinical examination, he had swelling on the left thigh and increase in size compared to the contralateral one, therefore was admitted for studying. Imaging findings including plain radiographs, CT, MRI and bone scintigraphy, as well as treatment performed, are described. To the best of our knowledge, is the youngest case reported in the literature.
La miositis osificante es un trastorno benigno caracterizado por la formación de hueso heterotópico en el músculo esquelético o en tejidos blandos. Es extremadamente rara en niños, <1% de los casos ocurren en menores de 10 años. Presentamos el caso de un niño de 17 días que, tras 10 días de ingreso en una Unidad de Cuidados Intermedios, fue derivado a nuestro hospital por una displasia del desarrollo de la cadera. En el examen clínico presentaba tumefacción y aumento del tamaño del muslo izquierdo respecto al contralateral, por lo que ingresó para estudio. Se describen los hallazgos en las imágenes de radiografías simples, tomografía computarizada, resonancia magnética y gammagrafía ósea; así como el tratamiento realizado. Hasta donde sabemos, es el caso más joven reportado en la literatura.
Ossifying myositis (OM) is a benign disease that consists of the formation of heterotopic bone in the muscles or soft tissues.1 This entity is clearly defined, and it can be divided into three types: progressive ossifying myositis (fibrodysplasia ossificans progressiva [FOP]), circumscribed or post- traumatic myositis ossificans (TMO) and atraumatic or pseudomalign myositis ossificans (PMO).2 The aetiology of the latter form is not fully understood at the current time. Although any part of the body may be affected, the anterior thigh is the most frequent site of appearance.3 In the primary stages O M lesions may present in a similar way to a rhabdomyosarcoma, osteosarcoma o lymphoma.1 It usually affects adolescents and young adults, with a slight predominance of the male sex.2,3
The case of a 17 days old infant with OM is presented, after a short period of immobilisation. Four aspects made the diagnosis of OM highly challenging for this child. Firstly, OM is extremely rare in children under the age of 10 years. Few cases have been reported in the literature, and to date, there have been no reports of it in newly-born infants. Secondly, the lack of known trauma meant that this diagnosis was not suspected. Nevertheless, although more or less recurrent trauma is found in 40%–60% of cases of OM, no trauma is present in many cases. Thirdly, the clinical presentation accompanied by other skeletal disorders and a peculiar phenotype were deceptive clinical signs and symptoms which could have placed it in the context of a syndrome. Lastly, the images obtained in different studies showed characteristics that are usually associated with musculoskeletal infections and malign tumours. When OM has a characteristic history and clear zonal pattern in imaging test results, diagnosis is relatively simple. However, it is not uncommon for the appearance of OM to suggest other considerations, which hinders diagnosis.
Clinical caseNew-born male after cephalic eutocic birth at 41 weeks, the result of the third pregnancy of a couple unrelated by blood, with one clinically healthy child and one dead at birth. The pregnancy had been monitored without incidents. The infant had an appropriate weight for his gestational age, with an Apgar score of 9 after one minute and 10 after 5min. Dysmorphic facial features stood out on examination, including hypertelorism, micrognathism and low ear implantation; bilateral cryptorchidism, slight axial hypotonia, 40° hips flexion-abduction with a positive bilateral Ortolani sign and rocker bottom feet.
An ultrasound scan of both hips was requested, showing right hip subluxation and left hip luxation with less than 33% bone coverage. Treatment with a Pavlik harness commenced. Serological tests to rule out congenital infections (TORCH) and metabolic tests were normal. Chromosome study was performed without finding any anomalies in the total metaphases studied. Chromosome formula 46XY is consistent with the masculine phenotype. Cranial, cardiac and abdominal ultrasound scans were normal, with no pathological findings. Ophthalmological, cardiological and ear, nose and throat examinations were normal.
At 48h of life he was admitted to the intermediate care unit (NICU) due to fever, idiopathic jaundice, 10% weight loss and hypernatremia without other signs of dehydration. He required serum therapy during 48h with normalisation of analytical data and the need for ventilator support with oxygen to maintain normal saturations. In an analytical check an increase in acute phase reagents was detected, so that he was treated with empirical intravenous antibiotic therapy until negative haemocultures were achieved. After 10 days in the NICU his respiratory state gradually improved and fever and the need for oxygen disappeared. The analytical parameters of infection normalised, so that he was moved to the paediatric ward.
At 17 days of life he was referred to our hospital to evaluate the dysplasia in hip development and convex valgus flat feet. In the left hip examination a swelling in the root of the thigh stood out, with increased size in comparison with the contralateral thigh, so that he was admitted to complete studies.
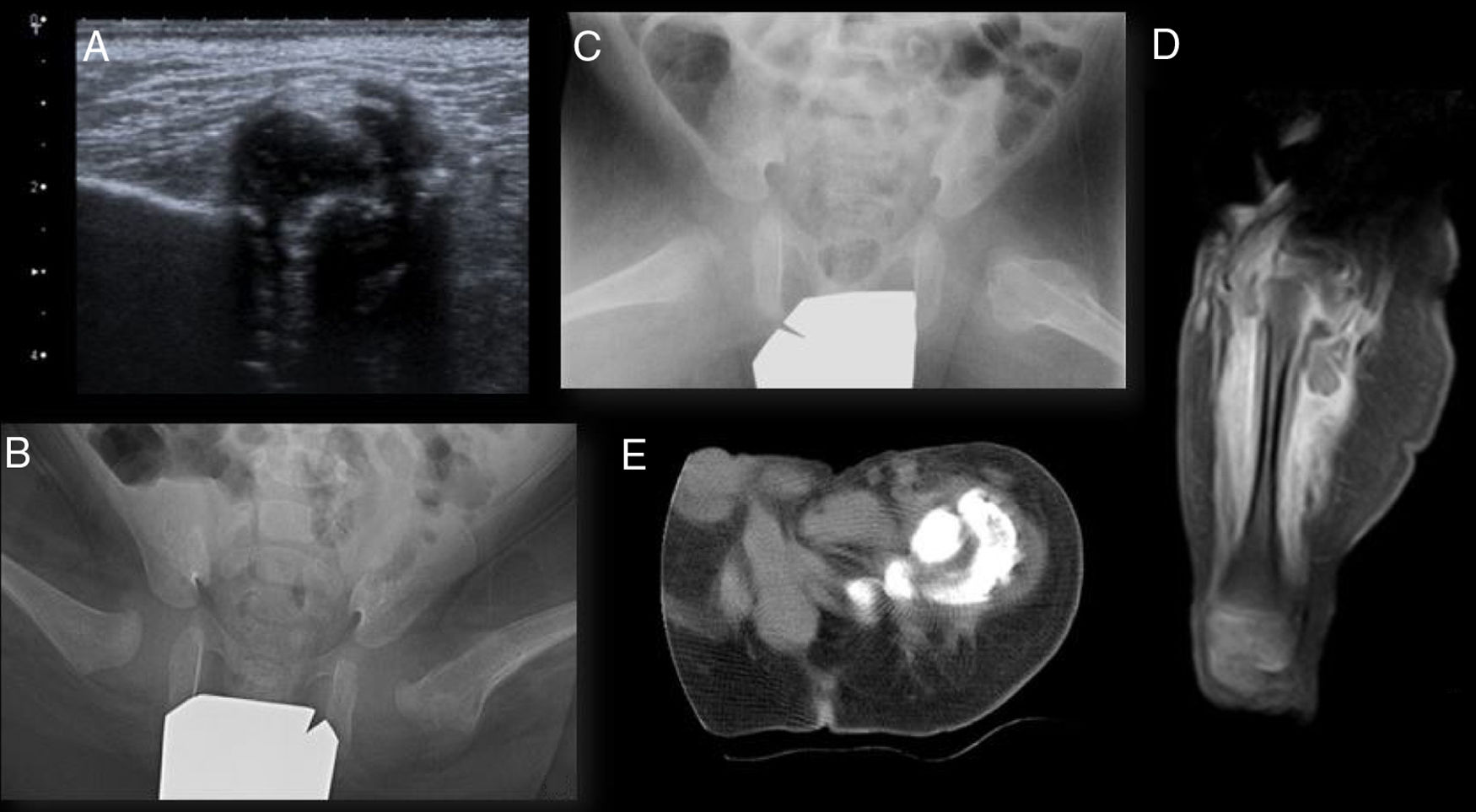
Ultrasound scan of the hips showed a large calcification inside the left gluteal muscles (Fig. 1a). Anteroposterior X-ray imaging of the pelvis showed luxation of the left hip and subluxation of the right hip, with an image with dense characteristics in the anteromedial zone of the proximal left femur; axial radiography of both hips also showed an alteration in the left proximal femur cortical bone (Figs. 1b and 1c). X-ray images of the feet showed bilateral vertical astragalus.
 Ultrasound scan of the left hip. Hyperechogenic image with a posterior acoustic shadow corresponding to a large calcification in the interior of the left gluteal muscles. (b) and (c) anteroposterior and lateral projection of the frog leg view of both hips. Increased density of the soft tissues adjacent to the cortex medial and lateral to the left femur head and neck, with cortical insufflation in the proximal femur. (d) Magnetic resonance image of the left hip. Continuous periosteal reaction in the femur with surrounding oedema and intramuscular calcification in the thickness of the muscles in the origin of the thigh. (e) CT of the left hip. Large calcification in the thickness of the muscles in the origin of the thigh, lateral, posterior, and medial to the femur, without being in contact with it. A bright zone between the lesion and the underlying bone, the presence of intact cortical bone, the location adjacent to the axis of a bone and the denser calcification in the lesion periphery are valuable radiographic findings for differential diagnosis vs bone malignity.")
Imaging tests. (a) Ultrasound scan of the left hip. Hyperechogenic image with a posterior acoustic shadow corresponding to a large calcification in the interior of the left gluteal muscles. (b) and (c) anteroposterior and lateral projection of the frog leg view of both hips. Increased density of the soft tissues adjacent to the cortex medial and lateral to the left femur head and neck, with cortical insufflation in the proximal femur. (d) Magnetic resonance image of the left hip. Continuous periosteal reaction in the femur with surrounding oedema and intramuscular calcification in the thickness of the muscles in the origin of the thigh. (e) CT of the left hip. Large calcification in the thickness of the muscles in the origin of the thigh, lateral, posterior, and medial to the femur, without being in contact with it. A bright zone between the lesion and the underlying bone, the presence of intact cortical bone, the location adjacent to the axis of a bone and the denser calcification in the lesion periphery are valuable radiographic findings for differential diagnosis vs bone malignity.
Magnetic resonance (MR) images showed infiltration of the muscles in the root of the left thigh associated with prominent intramuscular calcification, and a continuous periosteal reaction in the femur (Fig. 1d) with a surrounding oedema. Bone scintigraphy with 99Tc-diphosphonate showed intense osteoblastic activity in the region of the greater trochanter of the proximal left femur, without detecting other peripheral foci. The axial and coronal computed tomography (CT) reconstruction images of the pelvis showed an increase in the volume of the total thigh muscles, especially the quadriceps, semitendinosus and semimembranosus muscles, with a large calcification in the thickness of the muscles in the root of the same, lateral, posterior and medial to the femur, without being in contact with it, and a continuous periosteal reaction in the lateral and posterior region of the upper half of the femoral diaphysis (Fig. 1e). A percutaneous biopsy of the muscle tissue and bone was performed using endoscopy, and this had to be repeated in open form as it was not conclusive, with a definitive anatomical pathology diagnosis of OM.
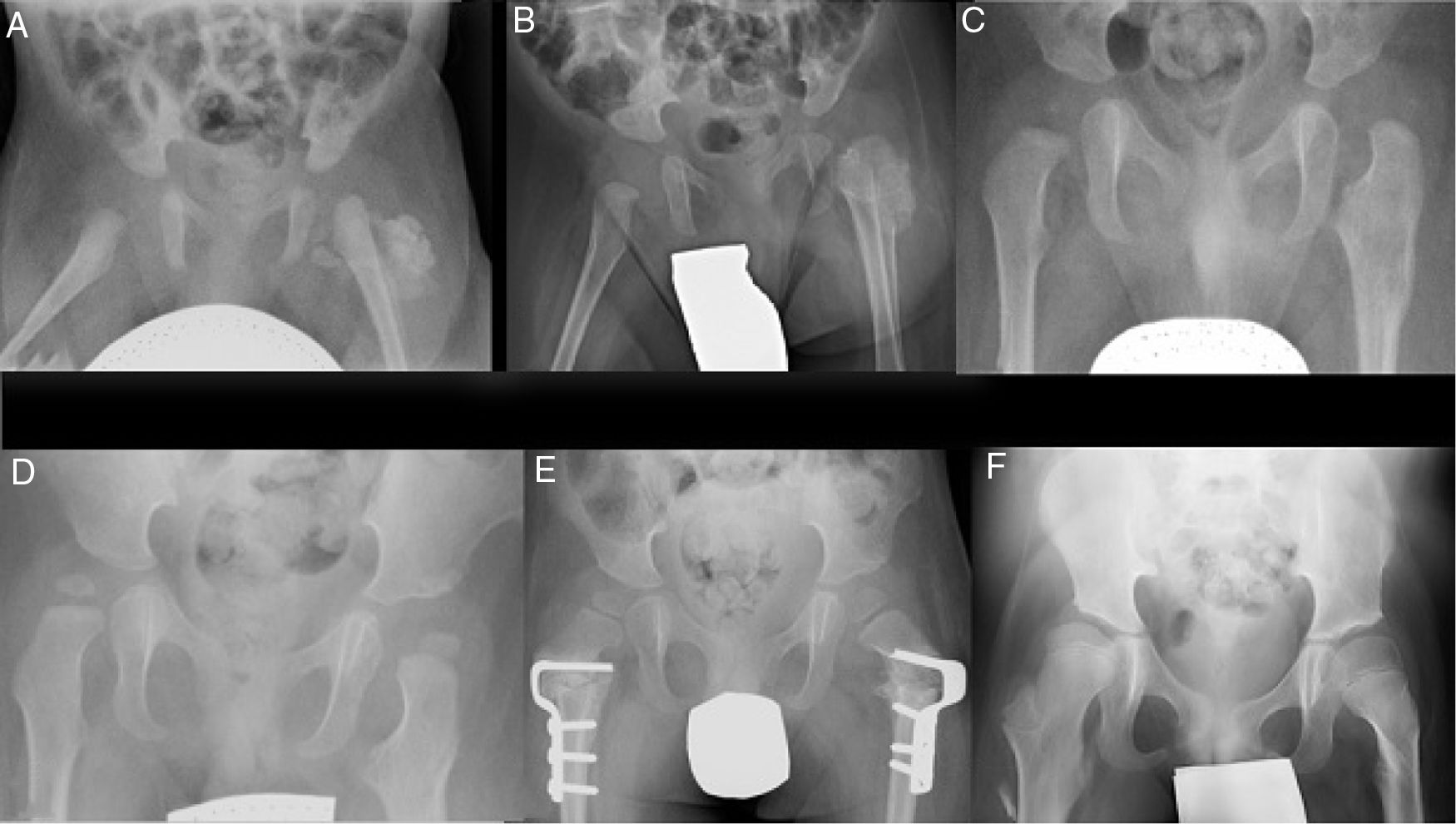
ResultsIt was decided to use conservative treatment closely monitored by series of conventional X-ray imaging tests (Fig. 2). These test showed a morphological alteration in the metaphysis as well as in the lateral cortical bone of the proximal left femur (Fig. 2c). This subsequently remodelled, together with a gradual reduction in the size of the ossified masses until they had been completely reabsorbed at 18 months (Fig. 2d).
 Evolution of the calcification in the thickness of the gluteal muscles and morphological changes at the level of the proximal femur at one month, 2 and 9 months, respectively. (d) Complete remodelling of the proximal femur at 18 months. (e) 3 years: bilateral proximal derotatory and modifying femoral osteotomy. (f) 8 years: hips reduced without evidence of recurrence of the lesion.")
Series of X-ray images during follow-up. (a–c) Evolution of the calcification in the thickness of the gluteal muscles and morphological changes at the level of the proximal femur at one month, 2 and 9 months, respectively. (d) Complete remodelling of the proximal femur at 18 months. (e) 3 years: bilateral proximal derotatory and modifying femoral osteotomy. (f) 8 years: hips reduced without evidence of recurrence of the lesion.
Due to his hip development dysplasia the patient was treated ineffectively with a Pavlik harness, and the left hip was stabilised by means of a closed reduction. The rocker bottom feet were treated with a series of plasters that were unable to reduce the deformity, so that corrective surgery was performed on both feet after one year of life, followed by treatment with an ankle orthosis. The patient underwent rehabilitation and psychomotricity therapy and was evaluated by Neurology at the age of 2 years because he was still unable to remain standing unassisted, even though the patient had worn plasters after the different surgical operations and tried to stand and to crawl. Due to the suspicion of delayed motor development he was referred to an Early Infancy Care Centre. At the age of 2 and a half years valgus of the proximal ends of both femurs was detected radiologically, in association with subluxation of both hips. Proximal bilateral femoral derotatory modifying osteotomy was therefore performed (Fig. 2e). At 5 years of age he started to walk with a frame, and at 6 years old commenced walking with sticks, requiring a wheelchair for long distances due to tiredness. The boy is now 8 years old and walks well with crutches, with plantigrade support of both feet, reduced hips and no evidence of recurrence of the ossified lesion at the level of the femur (Fig. 2f).
DiscussionOM is an uncommon entity that may appear at any age, although it rarely occurs in babies or elderly pacientes.3 The youngest patient reported in the literature was the case of a 5-month old girl, and the oldest was an 81 year-old woman.3 Our patient stands out for his young age and diagnosis at 17 days of life.
The most frequent locations for appearance of the lesion include the thighs, arms, shoulders and hands.4,5 It is rare to find it in the paravertebral region and the neck, and in the majority cases involvement is unilateral.1,4
Although pathogenesis in TMO is clearly defined, this is not the case in PMO, when the mass appears in the absence of trauma or evidence of systemic disease.3 In TMO it is presumed that initial trauma in a region caused a fibroblastic reaction, after which there was a certain degree of bone and cartilage metaplasia.2 In the majority of non-traumatic cases the following factors have been implicated as causal: repeated microtrauma or ischemic and inflammatory processes.3 Some authors argue that, particularly in young children, there was probably some type of trauma that went undetected.1,6 These hypotheses are also supported by the fact that no differences exist between TMO and PMO in terms of clinical presentation, images and histopathological changes. Both forms are therefore included within the general category of MO.6 Several studies have shown that OM is strongly associated with the expression of bone morphogenetic protein (BMP), which is more abundant in the plasma of children than it is in that of adults.7 It has therefore been reported that the mass matures and thereby ossifies more quickly in children than it does in adults, so that this permits a conservative approach to observing the typical maturing of a soft tissue lesion with the suspicion of OM in series of conventional X-ray images.6
Cases of PMO have been described after long periods of immobilisation due for example to burns, haemophilia, paraplegia, poliomyelitis, cranial lesions or prolonged hospitalisation.2 Only 2 cases have been reported in pre-school age children with no previous systemic pathology. These cases of OM occurred after prolonged admission to an ICU and both had bilateral involvement of the gluteal muscles.2 Our patient had unilateral involvement and a shorter time of admission in the NICU.
OM generally presents as a painful swiftly-growing mass without surrounding heat or erythema.1 in paediatric populations without a history of trauma the hereditary form of OM (FOP) should be considered, or OM secondary to child abuse.1,8 Progressive OM, which is also known as FOP or Münchmeyer’s disease, is a severe genetic disease that is extremely rare and disabling.1,8,9 It generally appears sporadically due to a genetic mutation, although it may also be inherited in dominant autosome form.1,8 FOP is characterised by progressive heterotopic connective tissue ossification. It commences in the first decade of life and all of the individuals affected have a congenital malformation of the big toes.1,8,9 Our patient had no external congenital anomalies of the big toes, thereby ruling out this diagnosis.
It is fundamental to know that OM generally progresses through 3 stages: the early stage, the intermediate stage and the mature stage.10 Each one of these stages is characterised by its own radiographic and histological findings and may hinder interpretation. Macroscopically OM has a structure similar to that of an egg, with a hard shell around it and a soft centre. At a microscopic level there is a proliferation of fibroblasts during the first week, with a high degree of mitotic activity, haemorrhaging and necrosis.5 In the second week this internal nucleus is surrounded by osteoblasts that produce immature osteoid bone, sometimes with cartilage.5 From the second to the fifth week these 2 zones remain delimited by an external layer of mature laminar bone, creating the zonal effect with 3 distinct zones.5 This zonal organisation is the characteristic histopathology of OM in the mature stage.5 As the lesion progresses, matures and calcifies from the periphery to the centre this centripetal pattern is the most important diagnostic characteristic in differentiating OM from malign tumours.10 Other factors which are also of use in differential diagnosis are the lack of invasion of the adjacent tissues and the inclusion of viable muscle fibres within the lesion.8,10 A biopsy is taken to definitively rule out a neoplastic process.2 It is important to take samples of all the elements of the OM, as in the initial stages if the biopsy is only taken from the central region, the mesenchymal cells which rapidly proliferate and have high mitotic activity may be diagnosed as malign.2,5
Analogously to what occurs at a histological level, simple X-ray images may be normal in the first stage due to a lack of mineralisation in the peripheral zone.3,10 During the intermediate stage it is possible to show there is peripheral calcification, while in a mature lesion diffuse calcification may appear as the osteoid mineralises.10 Calcifications appear in X-ray images at approximately 4 weeks.8 Evaluating the calcification pattern and bone involvement help to distinguish OM from other processes.
CT is the most precise imaging test, and it shows a smooth pattern of normal ossification within the thickness of a muscle, with clear separation from the bone.2 Depending on the ossification stage, the elements of laminar bone are found within the periphery of the ossified muscle, with a radiolucent central zone.2,7
MR images may be confused in the initial stages of OM, sharing many of the characteristics commonly found in musculoskeletal neoplasms and inflammatory processes, such as lesions with vague margins, heterogeneous signals due to the different cellular components of OM and the presence of diffuse oedema in the surrounding tissues.2,6 Weighted T1 images show a signal that is from iso- to hyperintense, and T2 weighted images show a hyperintense heterogeneous mass.10 In connection with the peritumoral oedema, this has been seen in children with musculoskeletal tumours in the same proportion around benign as well as malign lesions.6 Although it is rare, the periosteal reaction may be present in the OM, depending on its location and the topographical development of the lesion.6 With reference to this, OM has been classified in 3 types depending on its anatomical location: outside the bone (within the thickness of a muscle), parosteal (evolving in the immediate vicinity of or against a bone) and periosteal, which is also known as ossifying subperiosteal haematoma or periostoma.6,8 In periosteal and parosteal OM periosteal reactions may appear, and these may even simulate the so-called rising sun appearance that is typical of malign sarcomas.6 Our patient had an extensive muscle oedema as well as a continuous periosteal reaction in the femur; nevertheless, no cortical destruction or bone marrow abnormalities were found, and this once against rendered the diagnosis of malign sarcoma or infectious process improbable. It remains speculation as to whether these change reflect a reactive response, underlying inflammatory changes or simply bone contusion due to previous trauma.
Although bone scintigraphy is sensitive in the first phases of OM, it is not specific for diagnosis.2 It was useful in our patient to exclude multifocal lesions and the abused child syndrome.
Inflammatory parameters are often found to be raised in the blood of patients with OM, including ESR, CRP and alkaline phosphatase.1 Our patient had altered figures for CRP in the first hours of life, although we are unable to state that OM was the cause of this.
Differential diagnosis must include malign neoplasms as well as a range of benign lesions and infections. The presence of extensive muscle oedema as well as a continuous periosteal reaction in the femur may suggest the presence of a malign sarcoma such as Ewing’s sarcoma or osteosarcoma; however, both tumours calcify out from the centre towards the periphery, contrary to OM, and they adhere to the cortical bone on a broad base, usually invading the neighbouring tissues, with abnormalities in the bone marrow and cortical bone destruction.2,5 Osteosarcoma is extremely rare in children, particularly without previous irradiation.5 This lack of invasion of the adjacent tissues and the inclusion of viable muscle fibres in the lesion are useful in the histological differential diagnosis of extraskeletal, parosteal or synovial sarcomas.8,10 Rhabdomyosarcoma, which represents 50% of paediatric cases of soft tissue sarcoma, is found more often in the head and neck, the genitourinary tract and the limbs.5 The incidence of this sarcoma peaks during two ages: younger than 5 years and older than 15 years.5 In imaging tests localised bone erosion is often associated with the mass of soft tissues, and calcifications are infrequent.5 Synovial sarcoma should also be considered, but it is rare in children and approximately 30% of cases calcify.6 The said calcifications generally lack the peripheral border of ossification that is seen in OM, and they usually show highlights within the lesion posterior to the contrast.11,12 Chondrosarcoma too is a rare lesion in children.5 Soft tissue forms are exceptional, and moreover chondroid-type calcifications in these lesions have a dotted or ring and arc configuration.5 Other less frequent processes that it is important to distinguish from OM because they may cause soft tissue calcification include reactive periostitis when implanted in the bone surface and bizarre parosteal osteochondramatous proliferation (or Nora’s disease).12 Another soft tissue lesion which had to be differentiated is high grade undifferentiated pleomorphic bone sarcoma or malign fibrous histiocytoma, as it may have an osteocartiliganous component similar to MO.6
In the acute phase of OM, the appearance of MR may simulate a soft tissue abscess.12 Nevertheless, it classically has a hyperintense uniform appearance in sequences that are amplified in T2 and hypointense in T1, together with a periphery that stands out after the administration of contrast.12 In turn, CT with intravenous contrast shows a brilliant accumulation of liquid that emphasises the edge, confirming the suspicion of an abscess.12
Due to the patient’s phenotype and the coexistence of dysplasia in hip development and congenital bilateral vertical astragalus bones, the possibility of a bone dysplasia was raised; however, no nosological entity was found which could cover the set of clinical characteristics that had been observed. Melorheostosis was considered, a rare benign sclerotic bone dysplasia which has a sclerotomal distribution and is known to have a variant that is “similar to myositis”.12 The key factor in differentiating melorheostosis from OM is the identification of the sclerotomal pattern, which is no characteristics of MO.12 Neonatal cortical bone hyperostosis or Caffey’s disease was also considered, this being a rare and self-limiting inflammatory bone pathology that is almost exclusive to young infants. It causes fever and periosteal neoformation with thickening of the cortical bone and inflammation of the adjacent tissues, although it respects the epiphysis and metaphysis of the tubular bones11; in the same way pseudohypoparathyroidism was considered, together with its associated diseases, as it frequently presents with subcutaneous or progressive ectopic ossifications, although it also causes analytical alterations that were absent in our case.
Due to its rarity the principles for treating OM are based on empirical experience together with clinical or experimental evidence, and it basically consists of symptomatic treatment with non-steroid analgesic and anti-inflammatory drugs, accompanied by relative rest, given that the majority of lesions resolve within one year. Although indomethacin is used quite often in orthopaedics to prevent heterotopic ossifications, it has not been validated for the prevention and/or treatment of MO.13 No references to the use of glucocorticoids for this entity were found, and the literature does not recommend that they be used during the acute phase of a skeletal muscle lesion.13 When the clinical and radiological diagnosis is uncertain, or when the lesion causes pain, mechanical blockage of a joint or neurological deterioration, the mass is generally resected.3
To conclude, it should be underlined that OM is a rare entity in infants and exceptional in new-born ones. The diagnostic challenge is based on differentiating this lesion from malign bone and soft tissue tumours. The sensitivity of the imaging technique selected is determined by the stage of evolution of the lesion. Thus although there are characteristic images that suggest OM, they are not present until after the lesion has successively matured it develops the characteristic zonal pattern. CT is the imaging technique that shows, in the best way and at the earliest time, the typical ossification pattern in selected cases with suspected OM, so that it is able to confirm the diagnosis. MR imaging should be used only if the previous imaging techniques have not been able to establish the diagnosis of OM and a neoplasm is still suspected. A conservative approach has a definite role in this entity due to its self-limiting nature and spontaneous regression. Surgical treatment is reserved for lesions which cause functional restrictions or neurological deterioration. This case is of interest due to the extreme youth of the patient, his syndromic phenotype associated with other skeletal alterations, a short period of immobilisation, the lack of any reported trauma and the long follow-up period. The clinical characteristics of the patient make it possible to hypothesise a possible genetic aetiology, although no nosological diagnosis was established. In association with the latter point, we underline the importance of clinically following up the patient, as sometimes the clinical phenotype becomes fully defined with age; however, to date no new signs or symptoms have appeared that would suggest a nosological entity. No case of OM in children younger than 10 years described in the literature had previous pathology of interest, except one case of infantile cerebral palsy with OM in both hips.14 Two aetiological hypotheses are considered: firstly, pseudo-malign OM as a result of his admission to the NICU, and secondly, traumatic OM due to undetected trauma during birth. We consider that because of the unilateral nature of the lesion and the short duration of the patient’s immobilisation, a traumatic aetiology is more probable.
Level of evidenceLevel of evidence IV.
FinancingThis work was unfinanced.
Conflict of interestsThe authors have no conflict of interest to declare.
Please cite this article as: Lanuza Lagunilla L, Ramírez Barragán A, Miranda Gorozarri C. Miositis osificante en el lactante. A propósito de un caso. Rev Esp Cir Ortop Traumatol. 2021;65:152–157.
