





Folic acid and cobalamin are B-group vitamins that play an essential role in many cellular processes. Deficiency in one or both of these vitamins causes megaloblastic anaemia, a disease characterized by the presence of megaloblasts. Megaloblasts occur when inhibition of DNA synthesis causes asynchronous maturation between the nucleus and the cytoplasm. Clinical manifestations are similar to those of other types of anaemia, with the exception of cobalamin deficiency megaloblastic anaemia, which presents distinctive neurological symptoms. An understanding of the metabolism of these vitamins will enable clinicians to make the best use and interpretation of laboratory studies and monitor therapeutic strategies, which consist mainly of administering supplements to restore body reserves.
Anemias megaloblásticas: metabolismo del ácido fólico y vitamina B12 El ácido fólico y la cobalamina son vitaminas del complejo B, indispensables para un número importante de procesos celulares. El déficit de una o ambas vitaminas ocasiona anaemia megaloblástica, síndrome caracterizado por la presencia de megaloblastos, resultado de la asincronía de la maduración entre el núcleo y el citoplasma del eritrocito debido a la alteración en la síntesis de ADN. Las manifestaciones clínicas son similares a otras anemias, salvo la anaemia megaloblástica ocasionada por déficit de cobalamina que presenta alteraciones neurológicas de forma distintiva. Es importante conocer el metabolismo de las vitaminas en cuestión para un correcto uso e interpretación de los estudios de laboratorio, así como para el monitoreo de la terapéutica, basada esencialmente en reponer el déficit y restaurar las reservas corporales.
The discovery of megaloblastic anaemia and its aetiology was the result of the efforts of many different medical researchers. It was first characterized by Addison in 1849 as anaemia, general languor and debility.1 Osler and Gardner in 1877 noted the association with neuropathy, and 10 years later Lichtheim documented myelopathy. Megaloblasts were identified for the first time by Ehrlich in 1880. In 1920, abnormalities in white blood cells were described. In 1926, Minot and Murphy showed that the disease could be reversed by the intake of large quantities of liver.2 Three years later, Castle established that gastric acid contains an “intrinsic factor” that combines with an “extrinsic factor” to allow this latter to be absorbed.3 Hodgkin later identified the structure of vitamin B12, for which he received the Nobel prize.4 Years later, in 1948, Herbert discovered the structure of folic acid and described its association with the aetiology of megaloblastic anaemia.5
DefinitionMegaloblastic anaemia is a general term used to describe a group of anaemias caused by impaired DNA synthesis. It is characterized by abnormal findings in peripheral blood smear (macroovalocytes) and bone marrow samples (megaloblastic hyperplasia). Megaloblasts, the hallmark of these anaemias, are caused by asynchronous maturation between the nucleus and the cytoplasm due to DNA synthesis impairment.6–8
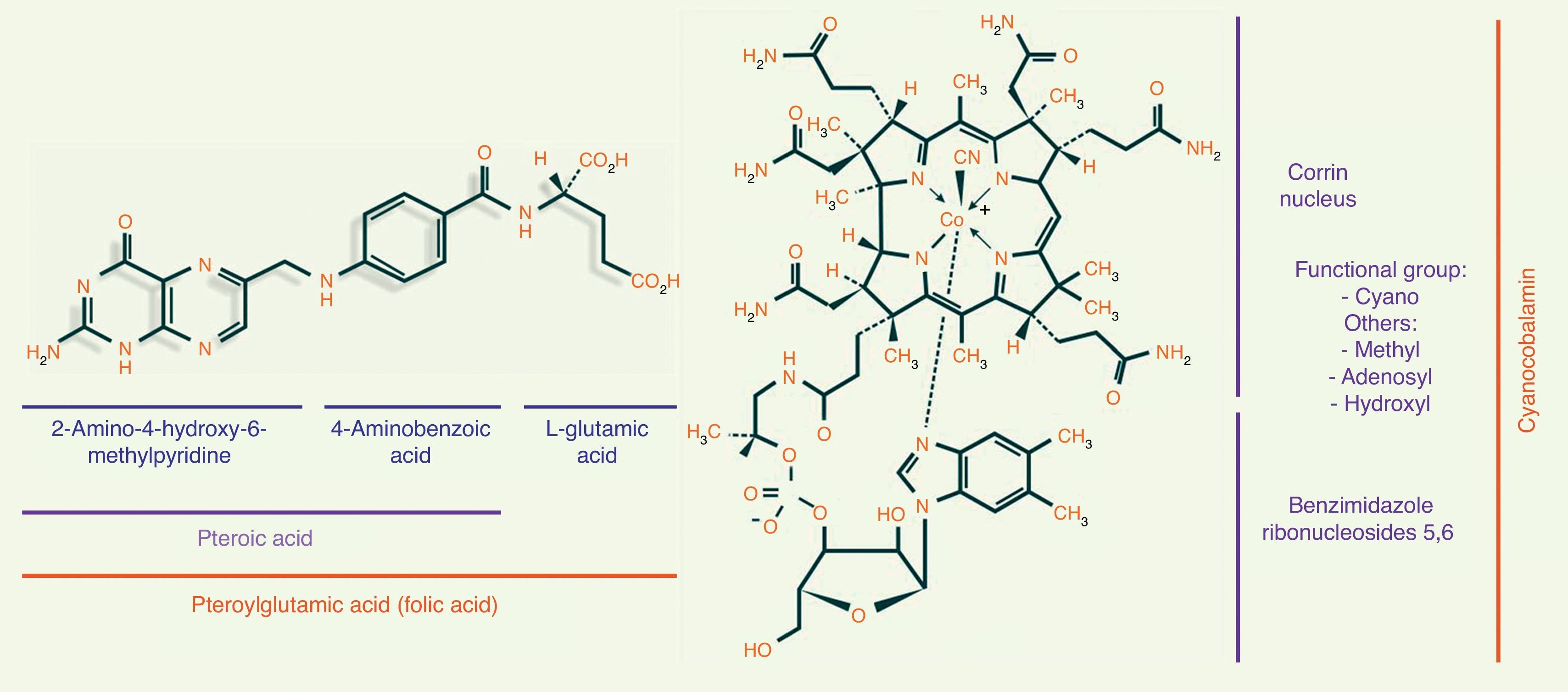
Folic acid metabolismFolic acid, also known as pteroyl-glutamate or pteroylglutamic acid, is made up of: (1) pteroic acid; and (2) l-glutamic acid (one or more strands) (see Fig. 1).8,9

The functional form of folate is tetrahydrofolic acid. The main dietary sources of folic acid are green vegetables, such as asparagus, broccoli, spinach and lettuce. It is also found in fruit, such as lemons, oranges, bananas and melons, and in cereals, grains, nuts, beans, beef, fish, liver and kidneys.6 Prolonged storage or over-cooking in abundant water can significantly reduce the folate content of food.10
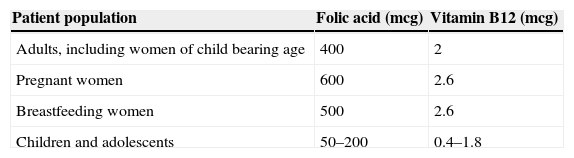
Daily adult requirements of folic acid range from 50 to 100mcg. The recommended dietary allowance (RDA) shown in Table 1, however, is far higher than the minimum requirement. This is because the bioavailability of folic acid depends on hydrolysation of the polyglutamate form of folate to its monoglutamate form to facilitate absorption across the small intestine.11
The body stores around 5mg of folate for between 3 and 4 months. Folate is primarily stored in the liver.12
Folic acid is mainly absorbed in the jejunum by means of passive transport following the concentration gradient, and by means of active transport when folate binds to reduced-folate transporter 1 and 2 (RFT-1 and RFT-2) and folate binding protein (FBP). Folic acid is also absorbed in the ilium, solely by passive transport.12–14 Folate, in either its monoglutamate or reduced monoglutamate form, is absorbed in a neutral pH environment (pH 7.4) facilitated by neutralization of the acid gastric environment by alkaline pancreatic juices.14 One of 2 enzymes, glutamate carboxypeptidase or polyglutamate hydrolase, are needed to hydrolyse folic acid polyglutamate (the form in which it is found in food) to its monoglutamate form. These enzymes are found on the luminal surface of the jejunum and ileum.13,14
Once taken up by the enterocyte, the enzyme dihydrofolate reductase mediates the conversion of folic acid to methyltetrahydrofolate through a two-step reaction. The folate then exits the enterocyte via the basolateral membrane and is either taken into the systemic circulation or the enterohepatic cycle (liver, bile acids, intestine). In the systemic circulation, 66% binds to albumin, 33% remains free, and a small amount (1%) binds to FBP. This is how it is transported to the cells where it will be used. It enters the cells either by binding to RTF-1 or RTF-2 or to folate receptor 1 (alpha) or 2 (beta). Intracellular folate transport is mediated by clathrin-mediated endocytosis. Once inside the cell (methyltetrahydrofolate), it must be demethylated to become tetrahydrofolate (functional folate that can accept glutamic acid chains; these in turn prevent it from exiting the cell, in other words, they “anchor” it inside the cell).13,15
Excess intracellular folic acid can pass into the blood stream and then be filtered through the glomerulus, secreted into the proximal tubule, and eliminated in the urine at a rate of 2–5mcg/day.16
The biological functions of folic acid include: serine-glycine conversion, histidine catabolism, purine synthesis, and more importantly, thymidylate and methionine synthesis.17
In thymidylate synthesis, folic acid carries one-carbon groups. Thymidylate is synthesized from deoxyuridine monophosphate (dUMP) and methylenetetrahydrofolate by thymidylate synthase, which converts these elements into dihydrofolate and thymidylate. Thymidylate, or thymine, is one of the 4 pyrimidine bases of DNA, and differentiates DNA from RNA (which includes thymine instead of uracil). In the absence of thymine, uracil is incorporated, thereby altering DNA synthesis.9,17
Metabolism of vitamin B12The chemical structure of cobalamin is shown in Fig. 1. Note the presence of 1 cobalt atom and 4 pyrole rings in the centre of the corrin ring. Cobalamin is given different names, depending on the radical to which it is bound. When it binds to a cyano radical, it is called cyanocobalamin or vitamin B12, a highly stable compound. Other functional forms of cobalamin include adenosylcobalamin (adenosyl radical) and methylcobalamin (methyl).
The main dietary sources of vitamin B12 are animal foods, such as beef, liver, fish, and dairy products. It is also found in some animals that ingest cobalamin-synthesizing bacteria, such as ruminants and oysters. Plant foods do not contain cobalamin.6,8
The recommended dietary allowance of vitamin B12 is shown in Table 1. It is important to note that the recommended intake reported in the literature ranges from 2 to 5mcg/day.9,11
The body stores between 2 to 5mg of vitamin B12 for between 3 and 4 months. Like folic acid, it is mainly stored in the liver.6,12,18
Cobalamin absorption in the ileum is mediated by a receptor called cubilin using a calcium-dependent passive transport mechanism. The cubilin receptor is actually a complex formed of cubilin and 2 proteins – megalin and AMN (the product of the amnionless gene, also involved in the production of amnion). It has a molecular weight of 460kDa, and is also found in the proximal tubule, where it mediates absorption of cobalamin itself.19 Absorption occurs in an acidic environment (pH 5.4).20 There are 3 cobalamin-binding proteins, which are also called cobalophilins; only 1 of these is a carrier:
- •
Transcobalamin I (TC I). Also known as haptocorrin or R protein. It is found in mature granulocytes and monocytes and also in precursor cells. It is also secreted by exocrine epithelial cells (found in the saliva, gastric acid, bile and breast milk). TC I binds to 70% of cobalamin and protects it from the acid environment of the digestive system. However, when it binds to cobalamin at other sites, it neutralizes the function of cobalamin.
- •
Transcobalamin II (TC II). TC II is synthesized by epithelial and endothelial cells, monocytes and fibroblasts. It binds around 30% of circulating cobalamin and is its only real carrier, transporting it to target cells where it will be used.
- •
Transcobalamin III (TC III). TC III is found in neutrophils; it has no known action. TC III levels are elevated in polycythaemia vera and other chronic myeloproliferative malignancies.21
Cobalamin is absorbed in the digestive system in three stages:
- •
Stomach. Cobalamin in food is bound to proteins from which it is released by the action of gastric acid and pepsin and rapidly taken up by TC I, which carries it to the duodenum.
- •
Duodenum and jejunum. The alkalizing action of the pancreatic juices together with the action of the pancreatic enzymes (tripsin, chymotrypsin and elastase) degrade the TC I and release the cobalamin, which is now taken up by the intrinsic factor (IF). IF is produced by the parietal cells of the fundus and cardia of the stomach. It protect the cobalamin and carries it to the cubilin in the ileum.
- •
Ileum. The IF-cobalamin complex binds to cubilin and is taken up into the enterocyte by means of a calcium-dependent passive transport mechanism.
Once inside the enterocyte, the IF-cobalamin complex is engulfed by lysosomes, where enzymes degrade the IF and release the cobalamin. The cobalamin then exits to the cytoplasm, where it is taken up by TC II. In this way, the TC II/cobalamin complex exits the enterocyte via its basolateral membrane and is released into the systemic circulation or the enterohepatic cycle (liver, bile acids, intestine). In the systemic circulation, cobalamin can bind to any of the 3 aforementioned cobalophilins. TC II transports it to the cells where it will be used. It is taken up into the cells by binding to either TC II receptors (TC IIR) or megalin (a protein). Cobalamin is taken up by cells by means of clathrin-mediated endocytosis, the same intracellular transport mechanisms used in folic acid uptake. Once inside the cells, TC II is degraded, thus releasing the cobalamin.13,15,22
Excess intracellular cobalamin can pass into the blood stream, from where it is filtered through the glomerulus and eliminated in the urine.19In humans, the 2 active forms of cobalamin involved in biological functions are:
- •
Methycobalamin. A co-enzyme of methionine synthase (also known as methyltetrahydrofolate-homocysteine methyltransferase), an enzyme involved in methionine and tetrahydrofolate synthesis from methyltetrahydrofolate and homocysteine. This is where the folate and cobalamin metabolism pathways meet, and it is sometimes called the “folate trap.”
- •
Adenosylcobalamin. A co-enzyme of methylmalonyl-CoA, an enzyme involved in the production of succinic acid from methylmalonyl acid (accumulation of which causes neuropathy). Succinate plays a role in the Krebs cycle, an energy-releasing pathway.
Two hypotheses have been developed to explain how cobalamin-deficiency anaemia is in fact caused by functional folate deficiency.23,24
- •
Methyltetrahydrofolate trapping, or the “folate trap”. Without cobalamin, methyltetrahydrofolate cannot be demethylated by methionine synthesis. Remember, methyltetrahydrofolate cannot be polyglutamised, and therefore cannot be “anchored” to the cell. This means that it can escape without being used.
- •
Formate deficiency. When tetrahydrofolate is depleted (as described above), stored methyltetrahydrofolate cannot be converted into polyglutamisable formate-mediated formyltetrahydrofolate, which is another functional form of folate (in addition to the oft-mentioned tetrahydrofolate) used in purine synthesis.
In addition to the foregoing hypotheses, cobalamin and folic acid metabolization share another common feature: they both require methylenetetrahydrofolate (a product of tetrahydrofolate) and dUMP to form thymidylate synthase-mediated thymidylate and dihydrofolate. For the reasons stated above, tetrahydrofolate cannot occur without cobalamin, and without tetrahydrofolate, neither methylenetetrahydrofolate nor its product, thymidylate, can occur.23,24
Pathophysiology of megaloblastic anemiaThe pathophysiology of this group of anaemias has its origins in ineffective erythropoiesis secondary to intramedullary apoptosis of hematopoietic precursor cells. This, in turn, is caused by DNA synthesis abnormalities.
Remember, both folate and cobalamin deficiency ultimately lead to thymidylate deficiency. DNA contains 2 purine bases (adenine and guanine) and 2 pyrimidine bases (thymine and cytosine).8,25
When there is insufficient thymidylate or thymine at the position in the DNA strand where these nitrogenous bases should occur, they are replaced by uracil. This happens primarily when uracil is incorporated at 2 similar positions in opposite strands. When uracil is incorporated into what should be a purely DNA structure, the repair enzymes detect the error and try to correct it, albeit unsuccessfully. As a result, first 1, then both DNA strands are destroyed, with the resulting p53-mediated cellular apoptosis.8,25,26
This in turn leads to asynchronous maturation between the nucleus and the cytoplasm. The latter, devoid of DNA, does not fully mature, and the former, in which RNA production continues and haemoglobin synthesis is unaltered, mature at the normal rate.8,9,25,26
Etiology of megaloblastic anemiaFolic acid deficiency is usually due to low folate content in the diet, or to an imbalance between folate demand and intake. Cobalamin deficiency is usually caused by poor absorption of this vitamin in the digestive tract (see Table 2).25,27
Causes of folic acid and cobalamin deficiency.
| Causes of folic acid deficiency | Causes of cobalamin deficiency |
|---|---|
| Dietary deficiency | Dietary deficiency |
| (a) Malnutrition(b) Elderly patients(c) Alcohol intake(d) Goat's milk intake | (a) Strict vegetarians |
| Malabsorption | Absorption problems |
| (a) Intestinal disorders. Tropical sprue, celiac disease, Crohn's disease, extensive small bowel resection, leukemic/lymphomatous infiltration, Whipple's disease, scleroderma, amyloidosis, diabetes mellitus.(b) Familial malabsorption. Intestinal conjugase deficiency Very rare disease. Inheritance: autosomal recessive Causes mental retardation, convulsions, ataxia or athetosis. | (a) Gastric disorders. Pernicious anaemia, gastritis, achlorhydria or hypochlorhydria (age, atrophic gastritis, proton pump inhibitors or H2 antagonists), Helicobacter pylori infection, total or partial gastrectomy, Zollinger–Ellison syndrome.(b) Intestinal disorders. Extensive ileal resection, regional ileitis, leukemic/lymphomatous infiltration, tuberculosis, Crohn's disease, postradiation ileitis, tropical sprue or celiac disease, scleroderma, amyloidosis, blind loop syndrome with bacterial overgrowth, Diphyllobothrium latum, Giardia lamblia, Strongiloides stercoralis, use of drugs that diminish absorption: cholestyramine, metformin, colchicine.(c) Pancreatic diseases. Hereditary chronic pancreatitis, Imerslund–Grasbeck disease (cubilin deficiency). |
| Genetic defects | Genetic defects |
| (a) Enzyme defects. Dihydrofolate reductase, methylenetetrahydrofolate and glutamate formiminotransferase deficiencies. | (a) Transcobalamin II deficiency.(b) Functional impairment of transcobalamin II.(c) Haptocorrin or transcobalamin I deficiency.(d) Homocystinuria.(e) Methylmalonic acidaemia.(f) Methylenetetrahydrofolate deficiency.(g) Cobalamin mutation. |
| Increased requirements | |
| (a) Pregnancy and breastfeeding.(b) Chronic haemolytic anaemia.(c) Chronic exfoliative dermatitis. | |
| Drug therapies | |
| (a) Trimethoprim. Inhibits dihydrofolate reductase.(b) Pyrimethamine. Inhibits dihydrofolate reductase.(c) Methotrexate. Inhibits dihydrofolate reductase.(d) Phenytoin and valproic acid. Reduce absorption and change its metabolism. | |
The clinical spectrum of megaloblastic anaemia is shown in Table 3, where the minor differences between the clinical manifestations of megaloblastic anaemia caused by folate deficiency and by cobalamin deficiency are highlighted.6,25,28
Clinical manifestation of megaloblastic anaemia.
| Clinical manifestation of megaloblastic anaemia |
|---|
| Folate/cobalamin deficiency |
| (a) Interview. Anaemic syndrome, sometimes tissue hypoxia.(b) Physical examination. Pale yellow colour (pallor/icterus), Hunter's glossitis, nail pigmentation, change of hair colour, splenomegaly (10–15%). |
| Cobalamin deficiency |
| (a) Neurological(1) Symptoms. Loss of joint position sense in the second toe, loss of vibration sense in toes and fingers, paraesthesia, hypoesthesia, tingling, gait abnormalities, loss of coordination, muscle weakness, spasticity, optic neuropathy, urinary and faecal incontinence, erectile dysfunction, dementia, memory loss. Neuropathy is symmetric, and mainly affects the lower extremities.(2) Signs. Positive for Romberg's test, Lhermitte's sign, Babinski reflex, spasticity, hyporeflexia, clonus.(b) Osteoporosis |
Cobalamin deficiency causes subacute combined degeneration of the posterior and lateral grey column of the spinal cord due to methionine deficiency. Methionine is needed for the production of myelin. Myelin deficiency causes demyelination and gliosis of the grey column, which is further aggravated by the neurotoxicity of methylmalonic acid. Elevation of tumour necrosis factor alpha and epidermal growth factor also contribute to neurological changes.28,29
Blood pictureAll types of megaloblastic anaemia, whether caused by folic acid or cobalamin deficiency, present the following laboratory findings.25
- •
Flow cytometry. In addition to anaemia, macrocytosis is found in 75% of cases (note that in 25% of patients mean corpuscular volume [MCV] is normal, above all in cases with concurrent iron deficiency or thalassemia). Macrocytosis can be classified as mild (100–105fL), moderate (106–115fL) or severe (>116fL). Another finding is increased blood cell distribution width. In some cases (associated with severe, chronic folic acid or cobalamin deficiency) varying degrees of leukoneutropaenia and thrombocytopenia can be present (usually mild to moderate, but occasionally severe).6,7,25
- •
Peripheral blood samples. The most notable findings are macrocytosis and hypersegmented neutrophils. From a morphological point of view, these abnormalities together, while not pathognomonic, are highly suggestive of megaloblastic anaemia. Hypersegmented neutrophils refers to the presence of >5% of neutrophils with 5 segments, or >1% with 6 segments. Other findings include anisocytosis, basophilic stippling, Howell–Jolly bodies, and giant hypersegmented granulocytes.7,25
- •
Reticulocyte count (percentage and absolute). Supravital staining shows reticulopaenia.6,25
- •
Biochemistry tests. These show ineffective haematopoiesis, characterized by intramedullary haemolysis, such as: elevated indirect bilirubin and lactate dehydrogenase (LDH). A certain amount of intravascular haemolysis can also be found, with reduced haptoglobin levels.6,25
- •
Bone marrow aspiration/bone biopsy. Megaloblastic changes occur in the morphology of the erythrocytic and myelocytic series. Red cell precursors (mainly orthochromatic erythroblasts) are enlarged and nucleus/cytoplasm maturation is asynchronous. This latter is characterized by immature nuclei (larger, with open or lax chromatin) and mature cytoplasm (with its normal red hemoglobulinised colour). Megaloblasts in the myelocytic series manifest as larger precursors (mainly bands). A less common finding is the presence of hyperdiploid megakaryocytes.6,7,25
- •
Other markers. Serum iron, ferritin and transferrin levels are elevated.6,25
Clinical presentation supported by common laboratory test findings usually strongly suggest megaloblastic anaemia. For specific diagnosis, however, folic acid and cobalamin levels must be quantified. Sometimes, quantification of intermediary metabolites such as methylmalonic acid and homocysteine may also be required. Table 4 shows the reference ranges and interpretation of these studies, together with a list of situation that can affect these results.6,7,25,30
Specific diagnostic tests for folate and cobalamin deficiency.
| Laboratory studies and diagnostic ranges | Situations affecting results |
|---|---|
| Serum folate | |
| <2ng/mL is diagnostic>4ng/mL rules out deficiency2–4ng/mL=quantify methylmalonic acid and homocysteine | Falsely low: Pregnancy, alcohol consumption, anti-seizure drugs, temporarily (a few days) deficient diet (with normal intra-enterocyte folate).Falsely elevated: Single intake of folate-rich food. |
| Intra-enterocyte folate (methyltetrahydrofolate [MTHF] and formyltetrahydrofolate (FTHF]) | |
| <100–160mg/L=deficiency63% of patients with cobalamin deficiency have low erythrocyte folate levels.Compared with serum folate, less likely to alter due to transient variations such as dietary changes. | Falsely low: Pregnancy, alcohol consumption, anti-seizure drugs, temporarily (a few days) deficient diet (with normal intra-enterocyte folate).Falsely elevated: Single intake of folate-rich food. |
| Serum cobalamin | |
| <200pg/mL diagnostic of deficiency>300pg/mL rules out deficiency in 95% of cases200–300pg/mL=quantify methylmalonic acid and homocysteineIntra-individual variation: up to 23% | Falsely low: Pregnancy, folate deficiency, HIV/Aids, anti-seizure drugs, multiply myeloma, hairy cell leukaemia, aplastic anaemia, myelodysplastic syndromes, paroxysmal nocturnal haemoglobinuria, Gaucher's disease, oral contraceptives, idiopathic origin, laboratory error. |
| Methylmalonic acid (MMA) | |
| Normal: 70–270mmol/LIntra-individual variation: up to 23%Usually elevated with comorbid cobalamin deficiency | Falsely elevated: Kidney failure, methylmalonic acidaemia.Falsely low: Use of antibiotics. |
| Homocysteine | |
| Normal: 5–14mmol/LIntra-individual variation: 17%Usually elevated with comorbid cobalamin and folate deficiency | Falsely elevated: Hereditary hyperhomocysteinaemia: changes in methyl-THFR, cystathionine beta-synthase, betaine synthesis. |
Elevated MMA and homocysteine: cobalamin deficiency (sensitivity: 94%, specificity: 99%).
Normal MMA and homocysteine: rules out deficiency of both vitamins.
Normal MMA and elevated homocysteine: folate deficiency (sensitivity: 86%, specificity: 99%).
It is important to bear in mind the possibility of subclinical cobalamin deficiency, found in 10–20% of geriatric patients. In this condition, serum cobalamin levels are slightly low, and AMM and homocysteine levels slightly elevated. In some cases, serum cobalamin, AMM and homocysteine levels can be within normal ranges. In the absence of clinical changes, and at times even in the absence of abnormal laboratory findings, this type of anaemia can only be diagnosed on the basis of high clinical suspicion.25,30
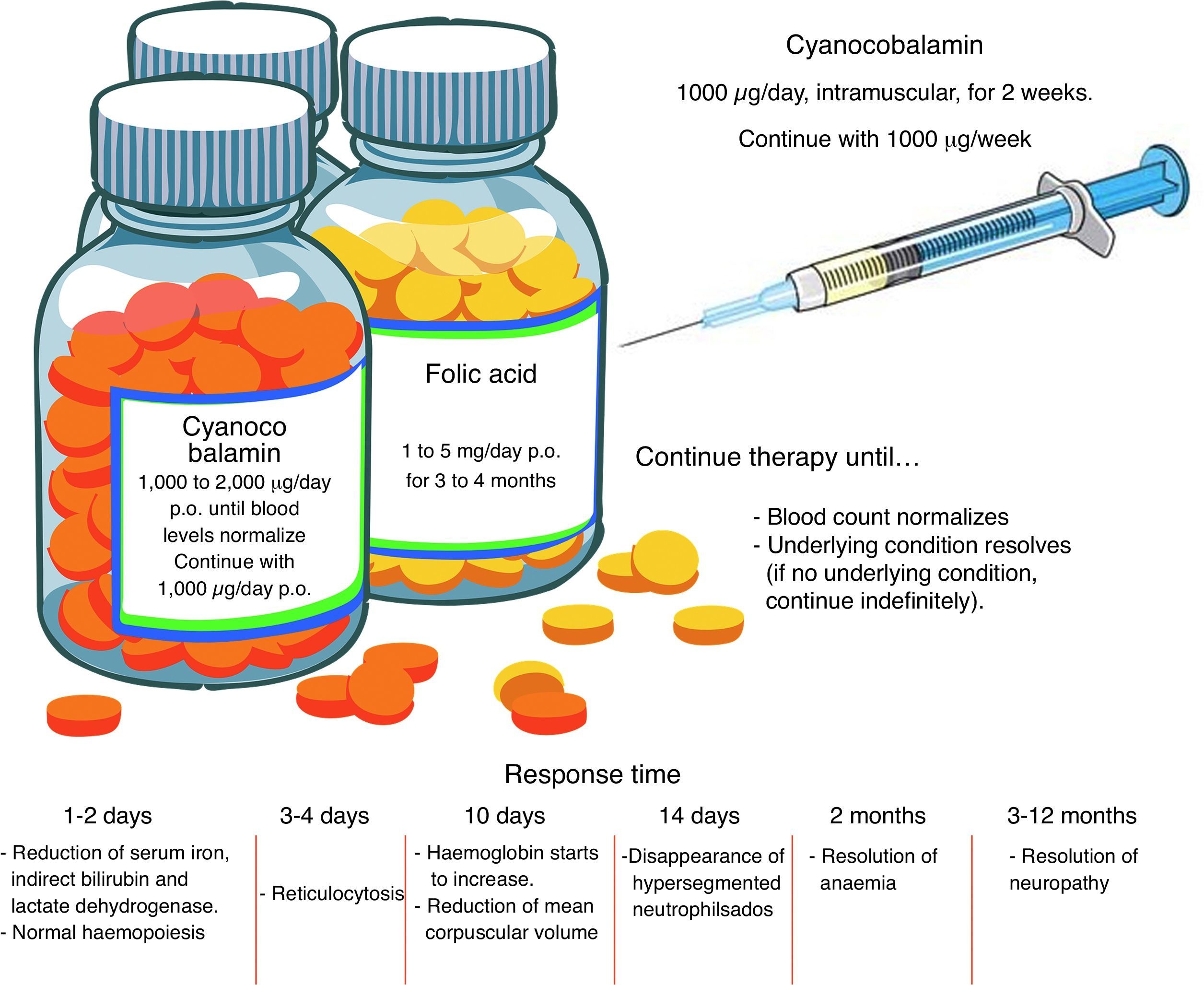
TreatmentFolic acid deficiencyBefore starting treatment for megaloblastic anaemia due to folic acid deficiency, it is important to ascertain the absence of concomitant cobalamin deficiency, and even to establish that cobalamin deficiency is not, in fact, the sole pathogenesis. When folates are given to a patient with cobalamin deficiency as the sole cause of the anaemia, or when both folic acid and cobalamin deficiency are involved, the blood picture will improve but neurological manifestations will worsen. High-dose, oral supplements should be given. See Fig. 2 for an overview of pharmacological management.11,23
Cobalamin deficiency
As in the case of folate deficiency, cobalamin deficiency therapy should continue until blood levels return to normal, or the underlying condition is resolved. The best route of administration is intramuscular. Excess cobalamin is excreted in the urine. Recent studies suggest that oral cobalamin is a safe and effective alternative, even in patients with low intrinsic factor levels. In patients that respond well to oral cobalamin, treatment should continue indefinitely at a dose of 1000mcg/day 9 Fig. 2).30–32
Clinical courseInitial response, in the form of normalization of haematopoiesis, is rapid. Reversal of neurological changes, however, takes longer (Fig. 2).8,12,25
Cobalamin deficiency prophylaxisIn certain patients, cobalamin supplements should be considered as part of routine clinical practice.
VegetariansStrict vegetarians should receive between 2 and 6mcg/day of oral supplement. Pregnant vegetarians (strict or not) that also intend to breastfeed need an even higher dose, as their offspring are at high risk of presenting severe cobalamin deficiency.33,34
Patients with prior gastric surgeryPatients with prior partial gastrectomy or gastric bypass surgery are at high risk for subclinical cobalamin deficiency or deficiency associated with impaired absorption of cobalamin. These patients should take 1000mcg/day cobalamin before meals.35,36
Elderly patientsIn the absence of well-designed studies investigating this topic, the benefit of routine administration of cobalamin supplements is unclear. In special circumstances, cobalamin levels can be quantified.6,37
Exposure to nitrous oxideNitrous oxide is known to inactivate cobalamin. For this reason, untreated or undiagnosed clinical cobalamin in patients scheduled for surgery using nitrous oxide may present rapid neuropsychiatric deterioration. The pre-operative workup of these patients, therefore, should include cobalamin tests or flow cytometry, and deficiency should be fully resolved before surgery. It is also important to bear in mind that nitrous oxide is sometimes used as a recreational drug, If chronic, this abuse can cause serious neuropsychiatric disorders, even when not associated with vitamin B12 deficiency.38,39
ConclusionsThe aim of this review has been to outline the essential information needed for the correct management of patients with megaloblastic anaemia. Cases not associated with a simple dietary deficiency, such as intestinal absorption disorders or cellular abnormalities, merit particular attention, as physicians not familiar with this aetiology will often have trouble diagnosing and treating these patients.
Pharmacological management appears to be straightforward. It is based supplementing deficits and building up body reserves. Follow-up of the latter is the key to a successful outcome in these patients. It is also important to consider the patient's dietary habits. Dietary advice should be given in the absence of an underlying medical condition preventing absorption of nutrients. In these cases, alternative strategies should be implemented to cover nutritional requirements.
Conflict of interestThe authors declare that they have no conflict of interests.
The chemical structure figures were taken and modified from the website Temas de Farmacognosia (http://www.plantas-medicinal-farmacognosia.com/gr%C3%A1fica/estructuras-de-las-vitaminas/).