


A síndrome poliglandular autoimune (SPGA) tipo II define-se pela presença de doença de Addison (DA) autoimune associada a Diabetes Mellitus tipo 1 (DM1) e/ou doença tiroideia autoimune. Habitualmente associa-se a outras doenças autoimunes «minor». Os autores apresentam o caso clínico de um adolescente com SPGA tipo II, manifestada aos doze anos de idade por DM1, hepatite autoimune tipo 1 com cirrose hepática e pancitopenia. No seguimento da investigação confirmou-se DA autoimune, presença de Ac antitiroideus, gastrite autoimune, doença celíaca, trombocitopenia imune e défice seletivo de IgA. No estudo da genotipagem do HLA-DQ foram identificados os haplótipos HLA-DQB1*03,*02. Inicialmente foi medicado com insulina, prednisolona e micofenolato de mofetil com resposta clínica parcial. Atualmente, quatro anos após o diagnóstico, encontra-se com dieta isenta de glúten, medicado com suplementos vitamínicos e minerais, insulina por bomba perfusora continua, prednisolona e azatioprina com controlo da função hepática e resolução da anemia, no entanto com difícil controlo glicémico. Os autores descrevem este caso clínico pela sua complexidade clínica e idade de apresentação, uma vez que esta síndrome é extremamente rara em idade pediátrica.
The autoimmune polyglandular syndrome (APS) type II is defined by the presence of autoimmune Addison's disease (AD) associated with type 1 diabetes mellitus (T1DM) and/or autoimmune thyroid disease. It is usually associated with other «minor» autoimmune manifestations. The authors report the case of an adolescent with APS type II, presented at the age of twelve by T1DM, autoimmune hepatitis type 1 with cirrhosis and pancytopenia. The clinical and analytical investigation confirmed autoimmune AD, presence of anti-thyroid antibodies, autoimmune gastritis, celiac disease, immune thrombocytopenia and selective IgA deficiency. During the study of genotyping of HLA-DQ the haplotypes HLA-DQB1*03,*02 were identified. Initially the patient was treated with insulin, prednisolone and mycophenolate mofetil with partial clinical response. Currently, four years after the diagnosis, the patient is on a gluten-free diet, vitamins and mineral supplements, insulin pump therapy, prednisolone and azathioprine with control of liver function and anemia resolution, but with difficult glycemic control. The authors describe this case for its clinical complexity and age of presentation, since this syndrome is extremely rare in paediatric age.
Introdução
A Diabetes Mellitus tipo I (DM1) resulta da destruição autoimune das células beta pancreáticas produtoras de insulina, provocada por uma interação complexa de fatores ambientais, genéticos e imunológicos1. A DM1 é caracterizada pelo aparecimento de insulite e presença de autoanticorpos (Ac) contra as células beta pancreáticas1. Em aproximadamente 33% dos doentes com DM1, a destruição autoimune não se limita ao pâncreas endócrino, desenvolvendo uma forma de síndrome poliglandular autoimune (SPGA)1-4. Dos doentes com DM1, 15 a 30% desenvolvem doença tiroideia (DT) autoimune (tiroidite de Hashimoto ou doença de Graves), 5 a 10% são diagnosticados com gastrite autoimune (GAI), 4 a 9% apresentam doença celíaca (DC) e 0,5% doença de Addison (DA)1-4. Estas doenças são caracterizadas pela presença de Acs contra a tiroglobulina e a peroxidase tiroideia (na tiroidite de Hashimoto), o recetor da TSH (na doença de Graves), a célula parietal (na GAI), a transglutaminase tecidual (na DC) e as células do córtex da suprarrenal e/ou a 21-hidroxilase (na DA)1-9. A deteção precoce destes Ac, marcadores de autoimunidade que se associam a infiltração linfocitária do órgão-alvo, são bons preditores de doença autoimune e precedem o défice hormonal e a doença clínica evidente4.
As SPGA caracterizam-se pela presença de duas ou mais doenças autoimunes endócrinas associadas a doenças autoimunes não endócrinas1-9. A SPGA autoimune tipo II, a forma mais comum das SPGA, é uma doença poligénica rara que afeta predominantemente mulheres em idade adulta1-9. Define-se pela presença de DA autoimune associada a DT autoimune (também designado síndrome de Schmidt) e/ou DM1 (designado síndrome de Carpenter)1-9. Habitualmente associa-se a outras doenças autoimunes endócrinas ou não1-9.
Uma vez que esta síndrome é extremamente rara na idade pediátrica, os autores descrevem o caso clínico de um adolescente com SPGA tipo II, manifestada aos 12 anos de idade, por DM1 e hepatite autoimune (HAI). A propósito do caso, fazem uma breve revisão teórica e tecem alguns comentários acerca dos aspetos práticos relevantes.
Descrição do caso clínico
Adolescente do sexo masculino, 17 anos de idade, raça caucasiana, com antecedentes perinatais irrelevantes. História de mononucleose infeciosa constatada aos cinco anos de idade e anemia microcítica hipocrómica desde essa data, de etiologia não esclarecida (sem investigação ou tratamento). Único filho, de pais jovens, saudáveis e não consanguíneos. História familiar (avós maternos) de DM tipo II, sem outros antecedentes familiares de relevo.
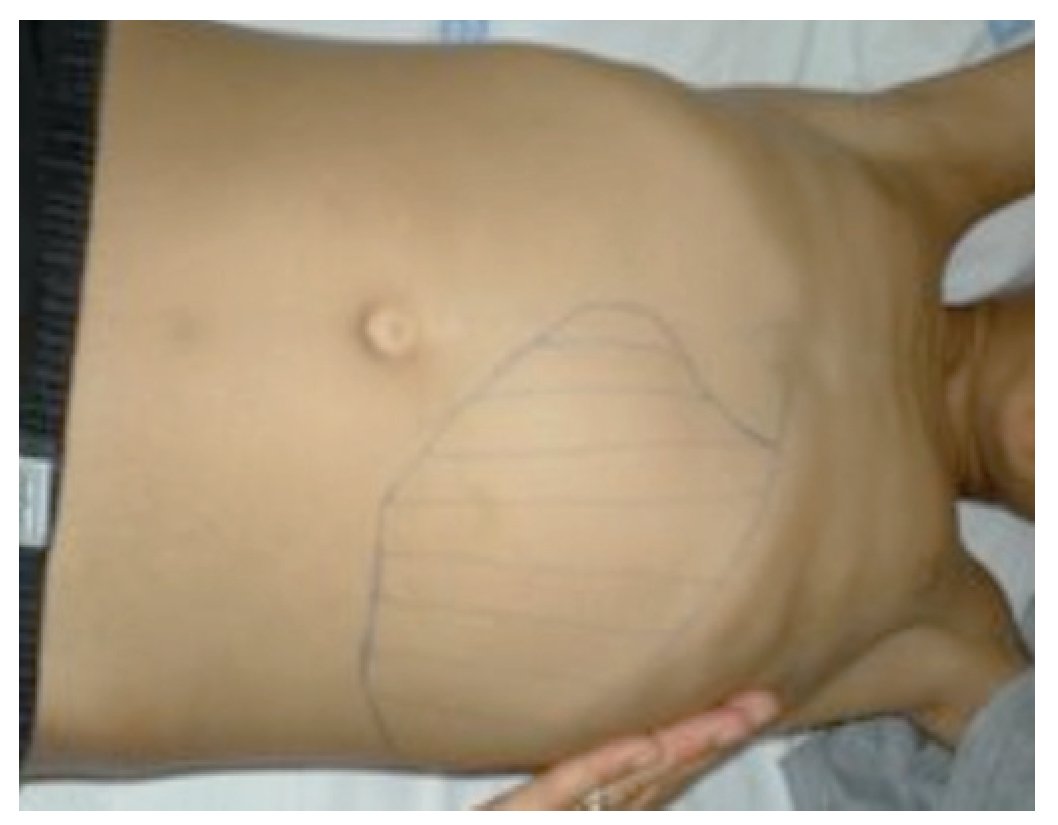
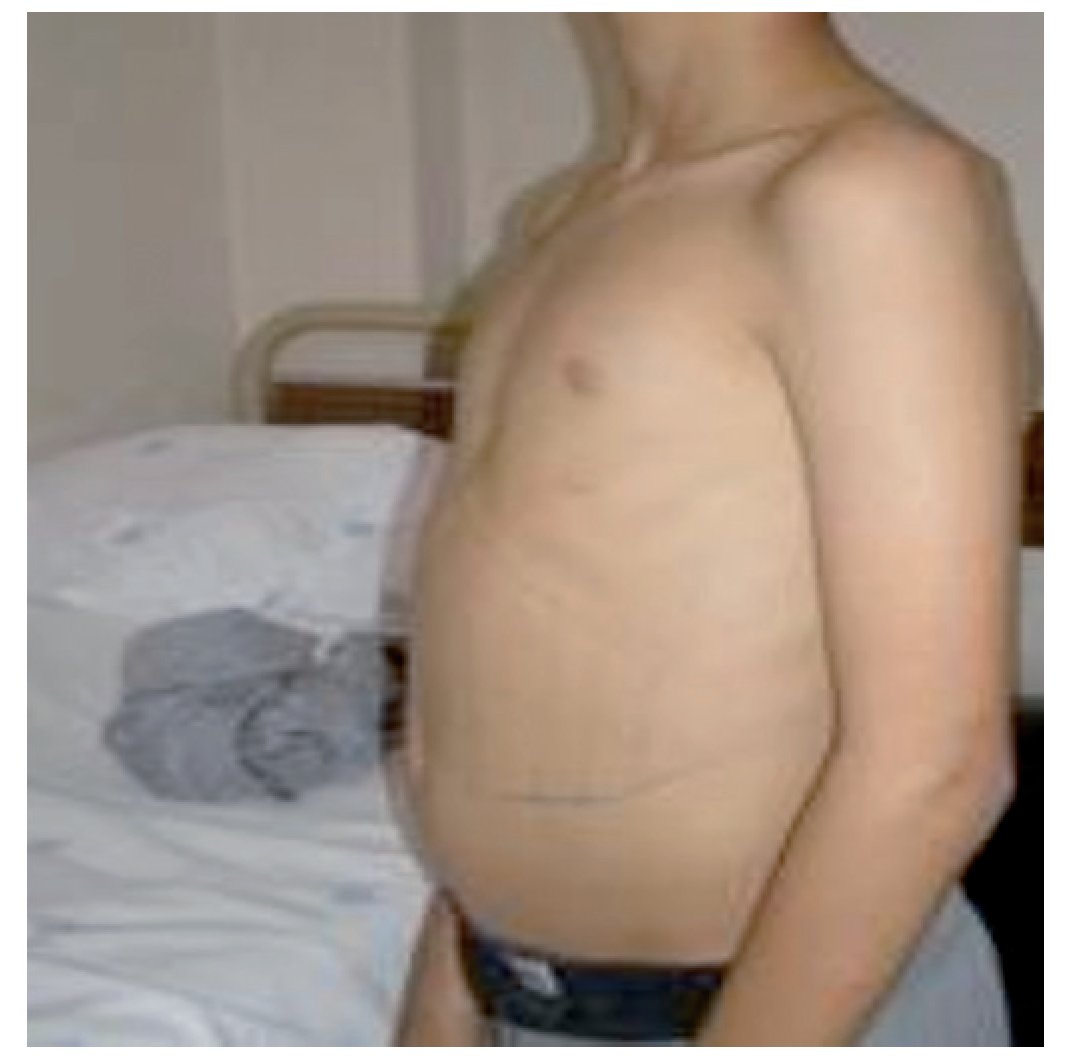
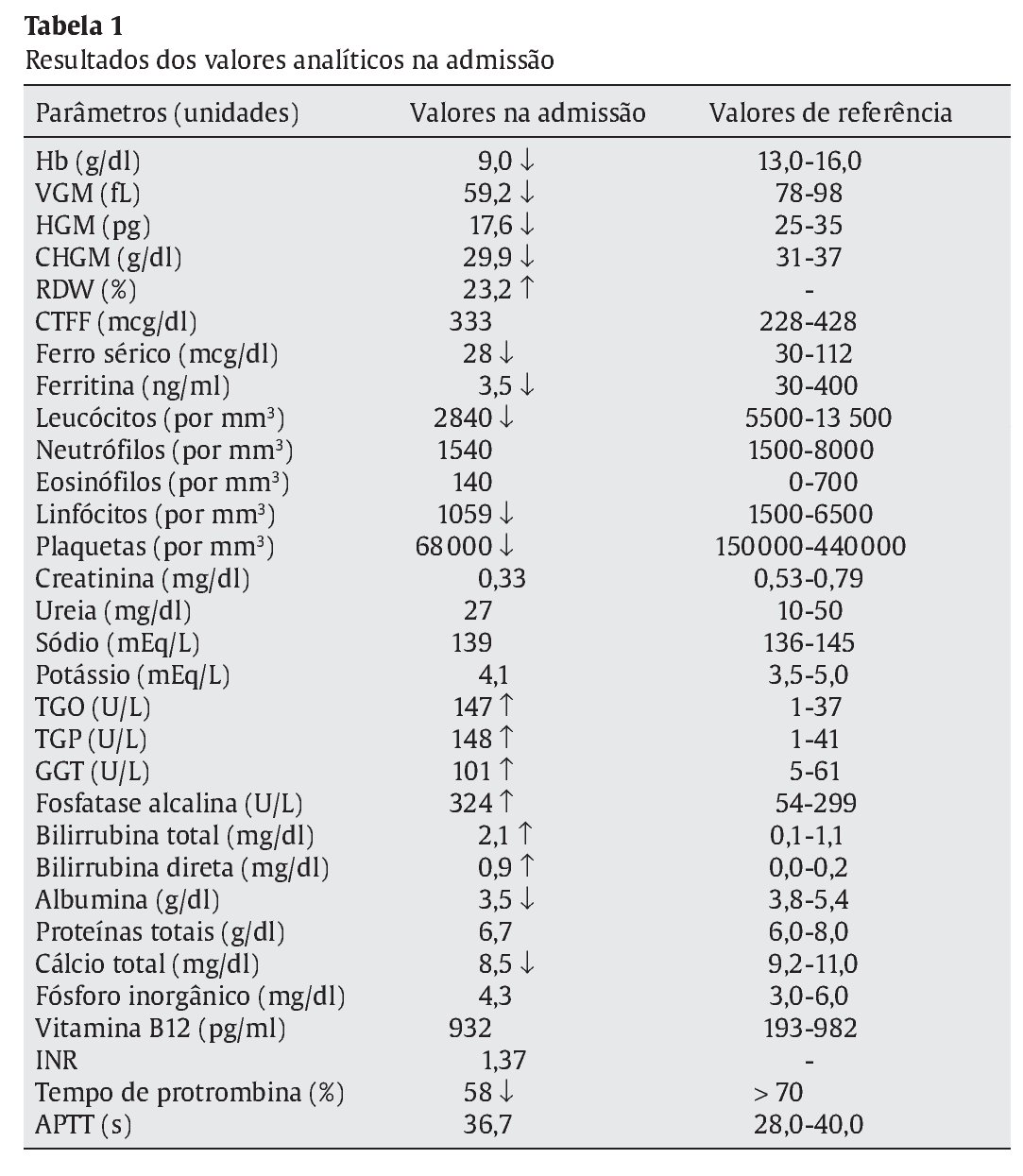
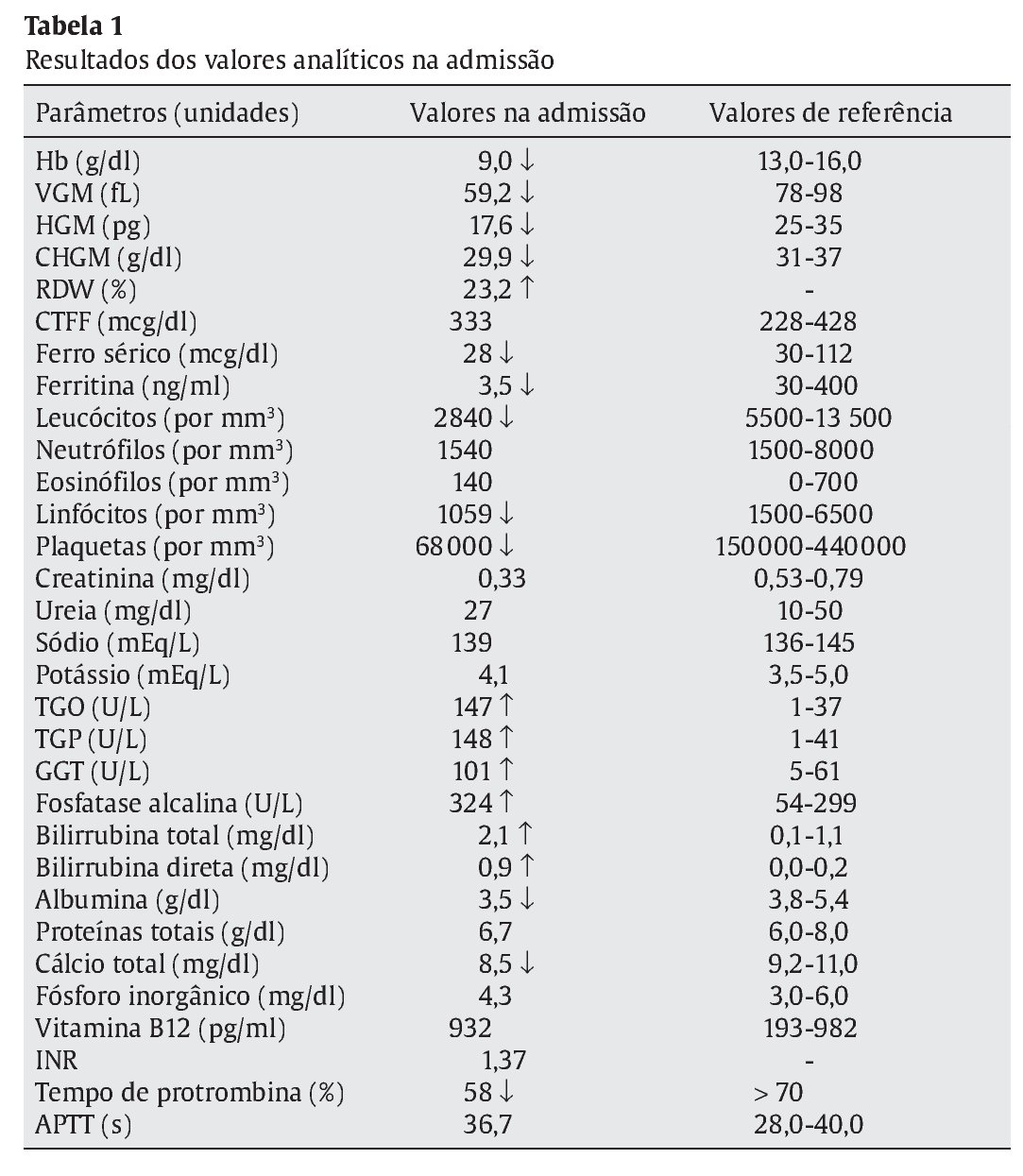
Referenciado ao serviço de urgência (SU) pediátrica aos doze anos de idade por quadro de polidipsia, poliúria, astenia e emagrecimento com três semanas de evolução. Ao exame físico apresentava aspeto emagrecido com perda ponderal de 10% (peso na admissão: 34,5 kg correspondendo ao percentil 5; estatura: 151 cm, no canal do percentil 25-50; índice de massa corporal: 14,9 kg/m2, no canal do percentil 3-5), tensão arterial 115/65 mmHg (canal do percentil 50-90), frequência cardíaca 89 bpm, hiperpigmentação cutânea generalizada, mucosas descoradas, desidratadas e subictéricas, abdómen globoso, doloroso à palpação do hipocôndrio direito e esplenomegalia (bordo esplénico palpável, 9 cm abaixo do bordo costal, ao nível da linha média clavicular esquerda, cruzando a linha média abdominal) (Figuras 1 e 2). No SU foi constatada hiperglicemia (267 mg/dl), glicosúria e cetonúria, sem acidose metabólica, com hemoglobina glicada (HbA1c) de 10,2%, insulina sérica de 2,8 uU/ ml (valores normais (N) 3,0-17,0) e peptídeo C de 0,97 ng/ml (N 1,1-4,4). O estudo analítico realizado à admissão também revelou pancitopenia com anemia microcítica e hipocrómica; ferropenia; hepatite com disfunção hepática e hipocalcémia (ver Tabela 1). Imagiologicamente, a ecografia abdominal com doppler revelou fígado de dimensões normais com ecoestrutura heterogénea, esplenomegalia homogénea (20 cm de diâmetro longitudinal), aumento do calibre da veia porta (13 mm), sem sinais de ascite ou alteração da permeabilidade dos vasos esplenoportais. No SU iniciou fluidoterapia, insulinoterapia e suplementos vitamínico-minerais (incluindo vitamina K e ferro oral), tendo sido internado para investigação diagnóstica e tratamento.
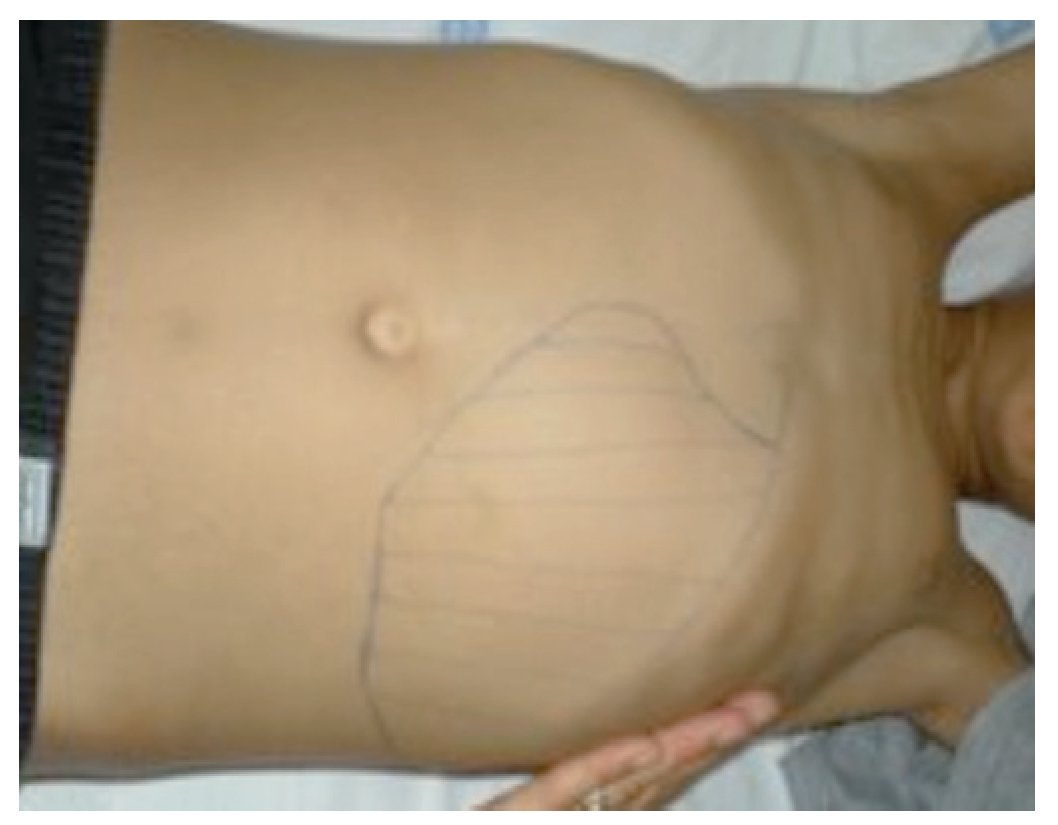
Figura 1. Esplenomegalia maciça.
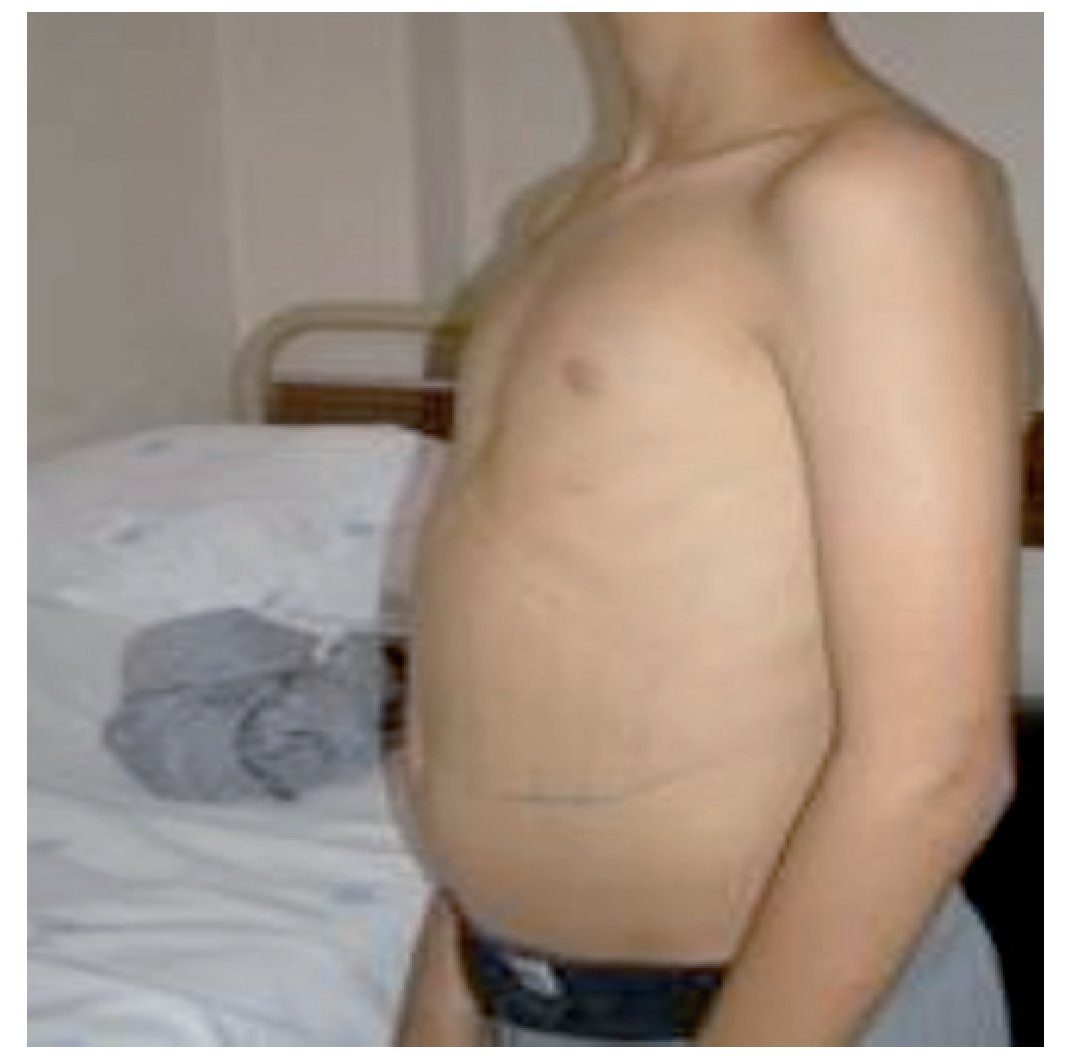
Figura 2. Esplenomegalia maciça.
No estudo imunológico detetou-se imunoglobulina (Ig) G sérica de 1500 mg/dl (N 621-1284), IgM sérica de 244 mg/dl (N 37-101), com doseamentos séricos normais da IgA e IgE, C3 52,0 mg/dl (N 75-135), C4 5,5 mg/dl (N 9-36) e positividade de inúmeros anticorpos (Acs) nomeadamente: antinucleares, antimúsculo liso, anticitoplasma de neutrófilo, anti-insulina, antiglutamato descarboxilase 65, anti-ilhéus de Langerhans, antiperoxidase tiroideia, antitiroglobulina, anticélulas do córtex suprarrenal, anti-21-hidroxilase, antiplaquetários, antigliadina, antitransglutaminase tecidual e anticélula parietal associados a gastrina de 199,7 pg/ml (N 0-108). A contagem dos linfócitos T CD3ab+CD4-CD8- foi de 1,5% (N < 2%) e das células T reguladoras (Treg) CD4+CD25+FOXP3+ de 10,5% (N 5-10%).
O estudo endocrinológico adicional revelou cortisol sérico matinal de 1,9 ug/dl (N 6,2-19,4) com valor sérico matinal normal da hormona adrenocorticotrófica (ACTH). Na prova de estimulação curta com 0,25 mg de ACTH detetou-se ausência de resposta do cortisol sérico (1,2 ug/dl) aos 60 minutos. O restante estudo endocrinológico não revelou alterações, nomeadamente o doseamento da paratormona (PTH), da função tiroideia e da testosterona total e livre. Nos estudos imagiológicos adicionais realizados não foram detetadas alterações morfológicas da tiroide e das glândulas suprarrenais.
Foi realizada biópsia hepática que confirmou hepatite crónica com fibrose portal e septal completa, com reação ductular com infiltrado inf lamatório mononuclear, hepatite de interface com infiltrado linfoplasmocitário e eosinofílico, transformação giganto-celular sincicial dos hepatócitos peri-portais e ligeira atividade necro-inflamatória com hiperplasia das células de Kupffer a nível lobular.
Na endoscopia digestiva alta (EDA) não foram detetadas varizes esofágicas ou gastropatia de hipertensão portal (HTP) e as biópsias gástricas e do intestino delgado, demonstraram gastrite atrófica crónica com infeção por Helicobacter pylori (Hp) e atrofia das vilosidades intestinais com infiltrado linfoplasmocitário da lâmina própria (classificação anatomopatológica de Marsh 3a), respetivamente. A colangioressonância magnética foi normal.
Foram excluídas etiologias infecciosas (hepatite A, B, C, VIH, Mycobacterium tuberculosis, Leshmania, citomegalovírus, vírus Epstein-Barr, e restantes infeções do grupo TORCH), neoplásicas (medulograma e biópsia óssea não revelaram alterações), metabólicas (doença de Gaucher I e doença de Niemann-Pick tipo B), a hemocromatose e a doença de Wilson.
A presença de DM1, DA autoimune e Ac antitiroideos permitiu o diagnóstico de SPGA tipo II. Associada a outras doenças autoimunes, nomeadamente hepatite crónica autoimune tipo 1, classificada segundo os critérios diagnósticos da HAI10 (pontuação igual a 21), com cirrose hepática e HTP; GAI com infeção por Helicobacter pylori (Hp); DC e trombocitopenia imune. No estudo de genotipagem do HLA-DQ foram identificados os haplótipos HL A-DQB1*03,*02. Não foram detetadas mutações no gene regulador autoimune (AIRE).
Durante o internamento foi mantida insulinoterapia (1,0 U/ kg/dia) e suplementos vitamínicos e minerais. Instituída dieta sem glúten, ferro endovenoso e terapêutica de erradicação do Hp com resolução progressiva da anemia. Após confirmação da hepatite autoimune foi iniciada terapêutica imunossupressora com prednisolona 40 mg/dia e micofenolato de mofetil 20 mg/kg/dia com resposta clínica parcial.
No seguimento multidisciplinar, foi constatada persistência da elevação das transaminases associada a discreta coagulopatia, pelo que a imunossupressão com micofenolato de mofetil foi alterada para a azatioprina (2 mg/kg/dia) associada a dose imunomoduladora de prednisolona com normalização da função hepática. No segundo ano de doença, iniciou insulina por bomba perfusora contínua com melhoria parcial do controlo metabólico da DM1 (diminuição da HbA1c média anual de 12,7% para 9,9%). No terceiro ano de seguimento, repetiu EDA verificando-se inexistência de varizes esofágicas, gastropatia de HTP, infeção por Hp e/ou alterações anatomopatológicas nas biópsias do intestino delgado. Por persistência da esplenomegalia maciça e trombocitopenia foi submetido a embolização parcial da artéria esplénica com diminuição do diâmetro longitudinal do baço e melhoria parcial da trombocitopenia, sem registo de intercorrências nomeadamente infecciosas. A partir do terceiro ano de doença, constatou-se défice seletivo de IgA persistente.
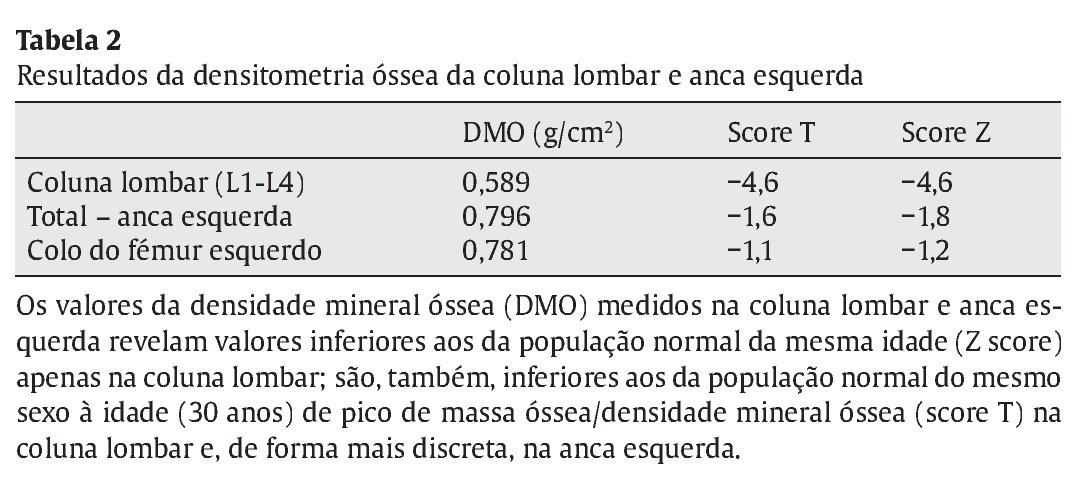
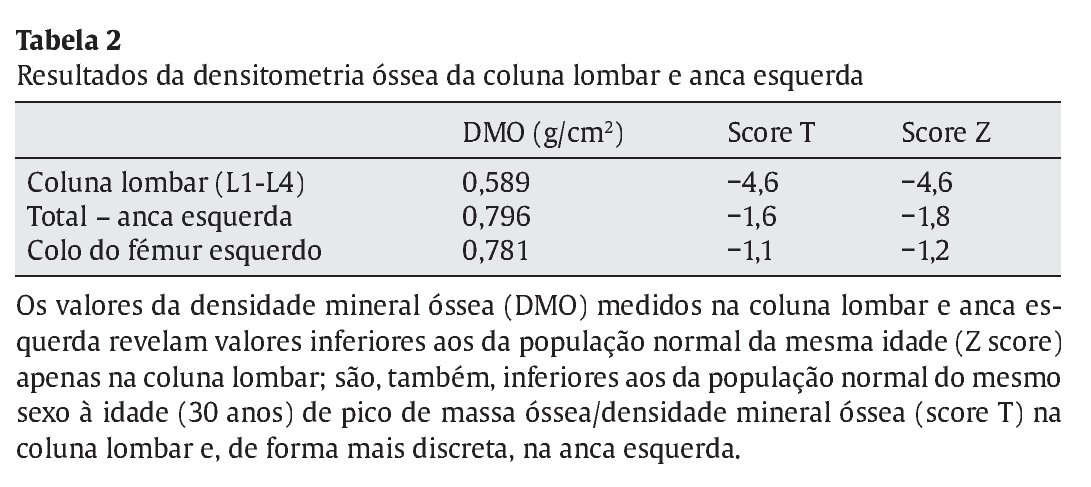
Em termos endócrino-metabólicos até ao momento, não foram diagnosticadas alterações da função tiroideia, dos valores séricos da P TH, complicações microvasculares da DM1 e/ou manifestações clínico-laboratoriais de insuficiência adrenal de novo. Manteve difícil otimização do controlo metabólico da DM1, devido à necessidade recorrente de doses imunomoduladoras de prednisolona, no entanto com desenvolvimento pubertário normal (atualmente no estádio pubertário grau V de Tanner). A cronicidade da patologia associada à imunossupressão crónica provocaram um atraso de crescimento com desaceleração estatural (estatura alvo familiar 171 cm, no canal do percentil 10-25; velocidade de crescimento 0,9 cm no último ano, no percentil 3; estatura atual 156 cm, inferior ao percentil 3, com atraso da idade óssea superior a 2 anos em relação à idade cronológica) associado a baixa densidade mineral óssea (ver Tabela 2).
Atualmente, quatro anos após o seguimento, mantém vigilância analítica regular, dieta sem glúten, suplementos vitamínicos e minerais (nomeadamente cálcio, vitamina D e ácido fólico), insulina por bomba perfusora contínua (1,6 UI/kg/dia), azatioprina (2 mg/kg/dia) e prednisolona (0,4 mg/kg/dia) em dose superior à dose de substituição preconizada para a DA, com HbA1c de 10,0% na última consulta.
O rastreio endócrino-metabólico e imunológico dos familiares em 1o grau permitiram o diagnóstico de tiroidite de Hashimoto no pai deste adolescente.
Comentário
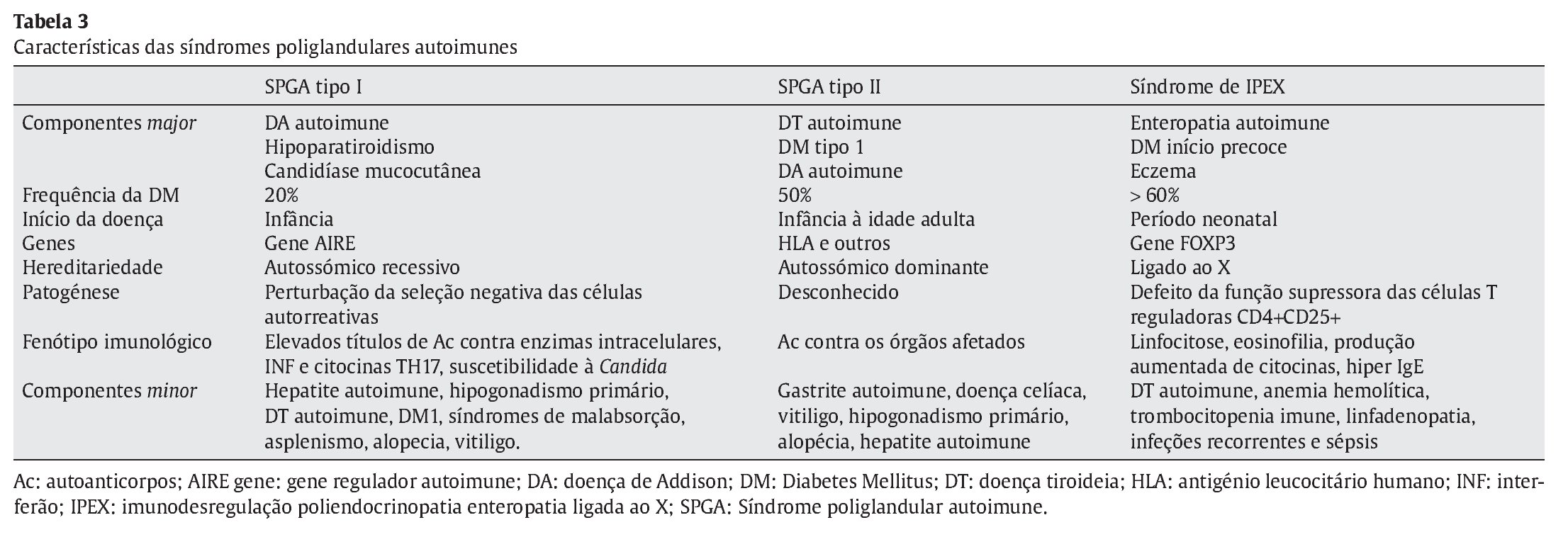
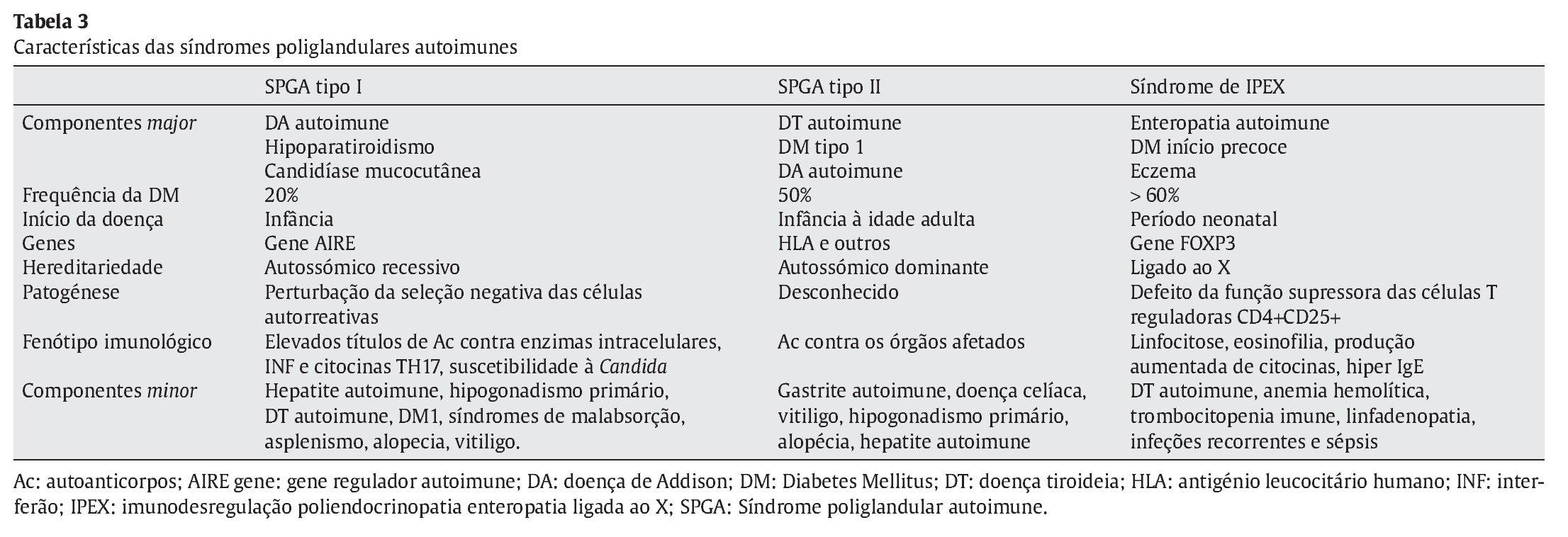
As SPGA caracterizam-se pela insuficiência funcional de duas ou mais glândulas endócrinas desencadeada por um processo autoimune1-9. O termo «poliglandular» nestas síndromes não é o mais correto, uma vez que também são descritas doenças autoimunes não endócrinas2,3. A maioria dos processos autoimunes destas patologias é associada a uma resposta celular T mediada, que resulta em destruição progressiva do órgão e produção de Ac órgãos específicos2,3. As SPGA têm sido classificadas de acordo com o defeito genético, padrão de hereditariedade, clínica e idade de apresentação1-9. Estas síndromes incluem doenças monogénicas, nomeadamente a SPGA tipo I ou APECED (autoimmune polyendocrinopathy candidiasis ectodermal dystrophy) e a síndrome IPEX, bem como outras doenças genéticas complexas, de que é exemplo a SPGA tipo II (ver Tabela 3)2,3. Em comum estas três SPGA apresentam um elevado risco de desenvolver DM1 (síndrome de IPEX > SPGA tipo II > SPGA tipo I)2,3.
Existem vários fatores implicados na perda de tolerância imunológica dos SPGA, nomeadamente genéticos, ambientais e imunológicos2-6,9. A autorreatividade das células T é determinada no timo (pela tolerância imunológica central) e na periferia, fortemente influenciada por alelos específicos do antigénio leucocitário humano (HLA)2-6. Postula-se que alguns agentes externos (como são exemplo os agentes infecciosos), que apresentem mimetismo antigénico, possam desencadear um processo autoimune2-6. De salientar neste caso clínico, a história prévia de mononucleose infecciosa.
A SPGA tipo II, a forma mais comum das SPGA, afeta preferencialmente indivíduos adultos jovens (pico de incidência entre os 20 e os 40 anos de idade) do sexo feminino (razão 3:1), com uma prevalência estimada de 1,4-2,0 em 100 000 habitantes1-9. Na literatura foram descritos menos de quinze casos clínicos com apresentação clínica igual ou inferior aos 12 anos de idade2-6.
É uma doença multifatorial, autossómica dominante com penetrância incompleta, com forte ligação aos haplótipos HLA classe II DR3 (DQB1*0201) e DR4 (DQB1*0302)1-9. Outros genes, como o polimorfismo do gene MIC-A (MHC class I chain-related A), do gene PTPN22 (protein-tyrosine phosphatase non-recetor type 22) e do gene CTLA-4 (cytotoxic T lymphocyte associated antigen-4) também foram associados a esta síndrome1-9. Neste doente foram identificados os haplótipos HLA-DQB1*03,*02, os quais conferem maior suscetibilidade para a DM1, DC, DA e tiroidite de Hashimoto1-9. O SPGA tipo II habitualmente está presente em várias gerações da mesma família, com apresentações clínicas diversas ou subclínicas2-6, como foi verificado no nosso caso clínico, no qual foi possível diagnosticar tiroidite de Hashimoto num familiar em primeiro grau.
Neste caso, o diagnóstico de SPGA tipo II, particularmente a síndrome de Carpenter, baseou-se na presença de DA autoimune e DM1, em associação à presença de Ac antitiroideus, sem alterações morfofuncionais da tiróide identificadas até ao momento1-9. Dada a descrição recente de casos clínicos atípicos com início tardio da síndrome de IPEX11, os autores pesquisaram o número de células Treg CD4+CD25+FOXP3+ nas células TCD4+, com o objetivo de rastrear esta patologia. A pesquisa de mutações no gene AIRE (típicas do SPGA tipo I) também foram investigadas, dada a apresentação clínica precoce, presença de DA autoimune em associação a hepatite autoimune (mais incidente no SPGA tipo I)12.
Na SPGA tipo II, a frequência relativa de cada endocrinopatia descrita na literatura é 100% para a doença de Addison, 69-82% para a disfunção tiroideia autoimune e 30-50% para a DM11-9. Apenas 10% dos doentes apresenta a tríade completa1-6. Em consonância com o nosso caso, a DM1 é a primeira manifestação clínica da SPGA tipo II em cerca de 50% dos doentes, diagnosticando-se posteriormente a DA autoimune e/ou a doença tiroideia autoimune4.
Associados aos componentes major do SPGA tipo II, são frequentemente repor tadas outras doenças autoimunes, nomeadamente a GAI (em 4,5 a 11%), hepatite autoimune (em 4%), DC (em 1 a 4%), dsIgA (menos de 1%) e trombocitopenia imune (menos de 1%) identificadas no nosso doente1-9. Dada a presença de inúmeros distúrbios autoimunes, nomeadamente a citopenia imune associada a hepatite autoimune e esplenomegalia maciça, os autores avaliaram os linfócitos T CD3ab+CD4-CD8- com o intuito de excluir a síndrome linfoproliferativa autoimune12.
Este caso clínico ilustra a importância de rastrear a DC e/ou GAI num doente com DM1 e anemia de etiologia não esclarecida1,2,4-8.
Os autores salientam ainda a importância do estudo da função suprarrenal e respetivos Ac órgãos específicos perante a presença de hiperpigmentação cutânea associada a sintomas inespecíficos (anorexia, astenia e perda ponderal) num doente com DM1, mesmo na ausência de hipoglicemia, hiponatrémia, hipercaliémia e/ ou acidose metabólica1-9. Neste caso clínico não foram descritas hipoglicemias de repetição, como frequentemente descrito na literatura em doentes com DM1 associado a DA e/ou DC1-9.
A terapêutica desta síndrome consiste na reposição hormonal de cada endocrinopatia integrada no SPGA tipo II, de forma semelhante ao que seria efetuado se estas fossem diagnosticadas isoladamente1-9.
Contudo a avaliação da função adrenal nestes doentes é essencial, antes de se iniciar tratamento com levotiroxina, a qual provoca uma depuração aumentada de cortisol, podendo evidenciar uma insuficiência adrenal subclínica pré-existente1-8. Os doentes com DM1 e DA autoimune habitualmente apresentam necessidade de reduzir as doses de insulina até que a insuficiência adrenal seja controlada, contudo se a dose de corticoide for muito elevada, as necessidades de insulina aumentarão exponencialmente1-8, tal como é descrito neste caso clínico. A abordagem terapêutica das doenças autoimunes não endócrinas integradas no SPGA tipo II é semelhante à preconizada se estas fossem diagnosticadas isoladamente1-9. Contudo sempre que possível, deverão ser selecionados tratamentos imunossupressores com menor incidência de efeitos laterais nas endocrinopatias associadas1-8.
Apesar da terapia hormonal de reposição continuar a ser o pilar de tratamento das SPGA, recentemente têm sido testados e utilizados em estudos experimentais, novos fármacos no tratamento do processo autoimune subjacente9. A DM1 tem sido a principal patologia endocrinológica, na qual se tem constatado maiores progressos nesta área. Os fármacos imunomoduladores têm como principal objetivo reverter o processo autoimune, consistindo em agentes antigénio-específicos e antigénio-inespecíficos. Estes últimos têm como alvo, vários componentes do sistema imune, incluindo os dirigidos contra as células T (anticorpos monoclonais anti-CD313, globulina antitimócito14 e ciclosporina15) e as células B (anticorpos monoclonais anti-CD2016,17). Acredita-se que os fármacos imunomoduladores antigénio-específicos induzam tolerância imunológica aos antígenos responsáveis pela destruição autoimune das células beta pancreáticas. Estas terapias incluem as vacinas com antiglutamato descarboxilase 6518, cadeia beta da insulina e péptidos semelhantes à insulina. Muitas destas terapêuticas reverteram a hiperglicemia em camundongos com DM1 de início recente, sendo uma promessa em seres humanos19. Noutras patologias endócrinas autoimunes, nomeadamente na Doença de Graves, tem sido utilizado o rituximab (anti-CD20), havendo estudos sugerindo alguma eficácia clínica nesta patologia com um bom perfil de segurança9,20,21.
Os autores salientam a importância do seguimento multi--disciplinar personalizado e vigilância clínico-laboratorial regular que permitem uma melhoria da qualidade de vida e eventualmente da sobrevida destes doentes.
*Autorparacorrespondência.
Correio electrónico:dianalexmoreira@gmail.com (D. Moreira).
INFORMAÇÃO DO ARTIGO
História do artigo:
Recebido a 31 de dezembro de 2011
Aceite a 4 de dezembro de 2012
