


O atraso pubertário representa uma causa frequente de referenciação à consulta de Endocrinologia Pediátrica. A sua etiologia é variada, classificando-se em hipogonadismo hipogonadotrófico ou hipergonadotrófico, consoante exista diminuição ou aumento das gonadotrofinas. A Síndrome de Kallmann é uma forma rara de hipogonadismo hipogonadotrófico e caracteriza-se pela sua associação a anosmia ou hiposmia. É causada por um defeito na migração dos neurónios que produzem a GnRH e dos que formam os nervos olfativos, cuja origem embriológica é comum. A doença afeta apenas a secreção de gonadotrofinas, sendo as restantes hormonas hipofisárias produzidas normalmente. Está frequentemente associada a outras malformações que podem ser um elemento fundamental para um diagnóstico precoce. Os autores apresentam três casos clínicos de Síndrome de Kallmann e malformações associadas.
Delayed puberty is a frequent cause of referral to pediatric endocrinologists. It may be due to a large number of causes divided in two major groups based on measurement of gonadotrophins: hypogonadotrophic or hypergonadotrophic hypogonadism. Kallmann syndrome is a rare form of hypogonadotrophic hypogonadism and is characteristically associated with anosmia or hyposmia. Its primary defect is an anomalous migration of the neurons responsible for the GnRH production and the neurons that compose the olfactory nerves. The syndrome only affects gonadotrophin production, while all other hypophysealhormones are normally secreted. In this paper we report and discuss three cases of Kallmann´s syndrome and associated anomalies.
Introdução
O atraso puber tário representa uma causa frequente de referenciação à consulta de Endocrinologia Pediátrica. Define-se como a ausência de desenvolvimento pubertário aos 13 anos na rapariga e volume testicular inferior a 4 ml aos 14 anos no rapaz1-4. São ainda critérios: amenorreia primária aos 16 anos ou intervalo superior a 2 anos entre o início pubertário e a menarca no sexo feminino ou intervalo superior a 5 anos até completar o desenvolvimento pubertário no sexo masculino2,4. A maioria dos indivíduos com atraso pubertário não tem patologia endócrina subjacente apresentando somente um atraso constitucional do crescimento e maturação. Contudo, um pequeno número de doentes pode apresentar uma causa patológica para este atraso e o seu diagnóstico é essencial já que podem necessitar de terapêutica específica3. De uma forma geral, as causas endócrinas de atraso pubertário podem dividir-se em dois grupos consoante o nível de gonadotrofinas (FSH e LH) seja baixo ou elevado: hipogonadismo hipogonadotrófico, de causa central, e hipogonadismo hiper--gonadotrófico, de causa gonadal, respetivamente1,5,6. Um dos grandes desafios prende-se com a distinção entre o hipogonadismo hipogonadotrófico e o atraso constitucional da maturação uma vez que não existe nenhuma prova que a consiga fazer de forma precisa3,6-8.
A Síndrome de Kallmann (SK), descrita pela primeira vez em 1856, é uma forma congénita de hipogonadismo hipogonadotrófico e caracteriza-se pela sua associação com um distúrbio olfativo (anosmia ou hiposmia)5,6,9-20. É uma causa rara de atraso pubertário, com uma incidência estimada de 1:10 000 a 1:80 000 e uma relação sexo masculino:feminino de 5:15-9,11,14,20. É causada por um defeito da migração neuronal que envolve as células produtoras de hormona libertadora de gonadotrofinas (GnRH) e os neurónios dos bolbos olfativos, cuja origem embriológica é comum6,10,11,14,17,19,21, com consequente produção insuficiente de GnRH e alterações do olfato. Cerca de 60% dos casos são esporádicos mas foram já identificadas três formas de transmissão: ligada ao X, autossómica dominante e autossómica recessiva5,8,9,11,12,14,16,17,19,21. Mutações em pelo menos 7 genes já foram descritas na SK - KAL1, FGFR1, PROK2, PROKR2, FGF8, CHD75,11,13 e WDR1121. De realçar que a mesma mutação em diferentes elementos da mesma família pode ter expressões fenotípicas muito variáveis6,13,15,16,21.
Os achados clínicos desta entidade incluem atraso pubertário, hábito eunucóide e diminuição/ausência da sensibilidade olfativa5,8,9,11-13,16,20,21. Do ponto de vista analítico, os níveis de gonadotrofinas (LH e FSH) estão normais ou diminuídos e os esteróides sexuais (estradiol e testosterona) estão anormalmente baixos para a idade, contrariamente às restantes hormonas hipofisárias que apresentam valores normais uma vez que a sua secreção não está afetada5,6,8,13,19,21. O estudo imagiológico da região hipotálamo-hipofisária é indispensável para exclusão de uma causa secundária de hipogonadismo hipogonadotrófico5,8,13,21. Na ressonância magnética (RM) cerebral, em 75 a 90%20 dos casos, são descritos hipoplasia do giro olfativo e aplasia ou hipoplasia dos bolbos e/ou tratos olfativos9,10,12-14. A sensibilidade olfativa pode ser aferida pela história clínica, embora a anosmia ou hiposmia sejam frequentemente subvalorizadas pelo doente5,11,13, por olfatometria ou pelo reconhecimento de odores particulares como o café13,21.
O estudo molecular é útil apresentando contudo uma sensibilidade de apenas 25 a 35%5,6,11,13,21. A descoberta de novos genes envolvidos nesta entidade ao longo dos anos tem aumentado a sensibilidade destes testes mas é provável que mutações em outros genes continuem desconhecidas. No sexo feminino, a ecografia pélvica é um método não invasivo que permite avaliar as características e dimensões dos genitais internos, que tipicamente têm configurações e dimensões pré-púberes4.
Tendo em conta que maioria dos indivíduos com SK não apresenta desenvolvimento puber tário adequado, um dos principais desafios consiste em induzir o processo de maturação sexual. Para este efeito, a administração de esteróides sexuais é habitualmente o tratamento de eleição4,5,8,13. No sexo feminino, o estradiol é administrado na forma transdérmica ou oral numa dose inicialmente baixa, procedendo-se a um aumento gradual da dose de forma a promover o desenvolvimento uterino e endometrial e otimizar o desenvolvimento mamário4,5,8,13. Após um período de 6 meses a 2 anos, ou antes se ocorrer a menarca, a associação de um progestagénio torna-se imperativa de forma a evitar a hiperplasia do endométrio4,5,9,13. Nesta fase, o tratamento passa a ser efetuado com anticoncecionais orais. No sexo masculino, o tratamento hormonal de substituição deve ser iniciado com baixas doses de testosterona intramuscular com aumento gradual da dose até atingir a dose do adulto (250 mg quinzenalmente ou a cada 3 a 4 semanas)3,5,8,9,13. Esta terapêutica hormonal de substituição deve ser mantida e ajustada, de forma a assegurar a função sexual, massa muscular, densidade óssea e massa eritrocitária adequadas5,13.
O tratamento da infertilidade pode mostrar-se mais complexo e nem sempre é eficaz3,4,9. No sexo feminino, a estimulação com GnRH pulsátil é utilizada para estimulação da foliculogénese5,13.
Este tratamento apresenta diversas vantagens em relação ao uso de gonadotrofinas uma vez que mimetiza o ciclo menstrual e diminui o risco de gestações múltiplas e síndrome de hiperestimulação ovárica5,13. Os dois tipos de tratamento apresentam taxas de conceção similares, que rondam os 30% por ciclo ovulatório5,13.
No sexo masculino, o sucesso para indução da espermatogénese ronda os 90 a 95%5,8,13. Para tal propósito usa-se tradicionalmente gonadotrofinas parentéricas (gonadotrofina coriónica humana associada ou não à FSH recombinante)5,8,13. No entanto, e uma vez que o defeito primário se localiza a nível da função hipotalâmica, a administração subcutânea pulsátil de GnRH, embora não aprovada para o efeito, tem-se mostrado ef icaz em estudos experimentais nos indivíduos que não respondem à terapêutica com gonadotrofinas5,8,13.
Na grande maioria dos casos, o déf ice de produção de gonadotrofinas é permanente. Contudo, em cerca de 10% indivíduos do sexo masculino8, foi descrita a reversão do quadro após tratamento mais ou menos prolongado de reposição hormonal12.
Este «despertar» do eixo hipotálamo-hipófiso-gonadal sugere que um estímulo externo, ainda desconhecido, possa estimular os neurónios produtores de GnRH, numa fase mais tardia5,12-16,21.
Casos clínicos
Caso 1
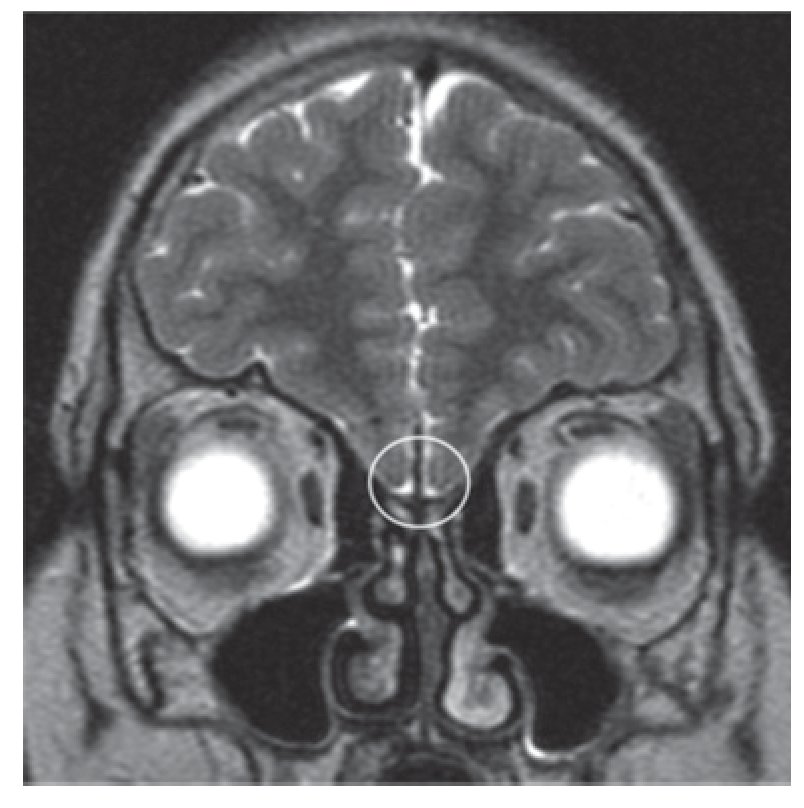
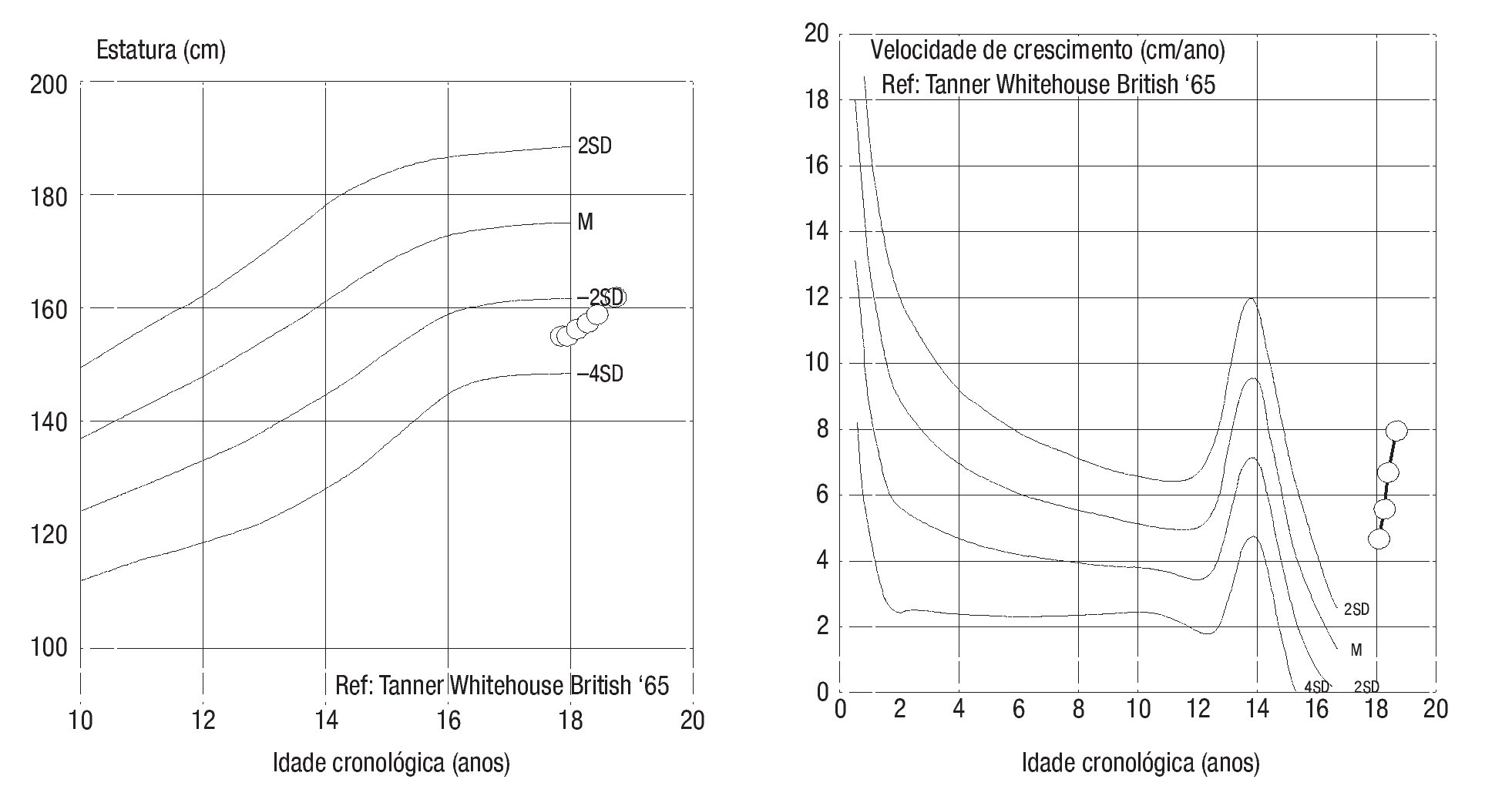
Adolescente do sexo masculino, enviado à consulta aos 17 anos e 10 meses por ausência de desenvolvimento dos carateres sexuais secundários. Relatava anosmia e tinha antecedentes de orquidopexia bilateral aos 16 anos. Os antecedentes familiares eram irrelevantes. Ao exame objetivo apresentava uma assimetria facial, um sopro sistólico grau II/VI audível no bordo esternal esquerdo, micropénis (comprimento do pénis de 6 cm), pilosidade púbica Tanner 1 e volume testicular inferior a 3 ml. Não apresentava pilosidade facial e a voz era de tonalidade aguda. A estatura era de 154,5 cm (desvio padrão (SD) -3,03 e percentil < 1) para uma estatura alvo de 167,3 cm. Do estudo realizado realça-se: idade óssea com atraso de 4 anos e 4 meses em relação à idade cronológica; IGF-1 de 135 ng/ml (Normal para a idade cronológica 136-780), função tiroideia e prolactina normais e gonadotrofinas e testosterona total baixas (LH: 0,1 mUI/ ml; FSH: 0,8 mUI/ml e testosterona total: 14,3 ng/dl). Efetuou prova de clonidina com priming com testosterona com pico sérico de hormona de crescimento de 7 ng/ml. A RM cerebral excluiu patologia do eixo hipotálamo-hipófise mas revelou: «...ausência dos bolbos olfativos e dos 2/3 anteriores dos sulcos olfativos..» (Figura 1). O estudo genético realizado no Centro de Genética Clínica identificou uma mutação no gene KAL1 - deleção do exão 1, c.67_92del26, p.Leu23fsX54 - nunca antes descrita. Efetuou ecocardiograma que não revelou alterações e ecografia reno-pélvica onde se detetou agenesia do rim direito. Iniciou testosterona intramuscular aos 17 anos e 10 meses em doses crescentes, com boa evolução clínica. Aos 19 anos (14 meses após início da terapêutica) apresentava pilosidade púbica Tanner 5, volume testicular de 6 ml e pénis com 10 cm de comprimento, bem como desenvolvimento de outros carateres sexuais secundários: barba e voz grave. A estatura era de 167,6 cm (SD -0,65; percentil 25), acima do previsto de acordo com o património genético, e a velocidade de crescimento de 5,1 cm/ano (SD 1,11; percentil 86 para a idade óssea) (Figura 2). Foi referenciado para a consulta de adultos para seguimento.
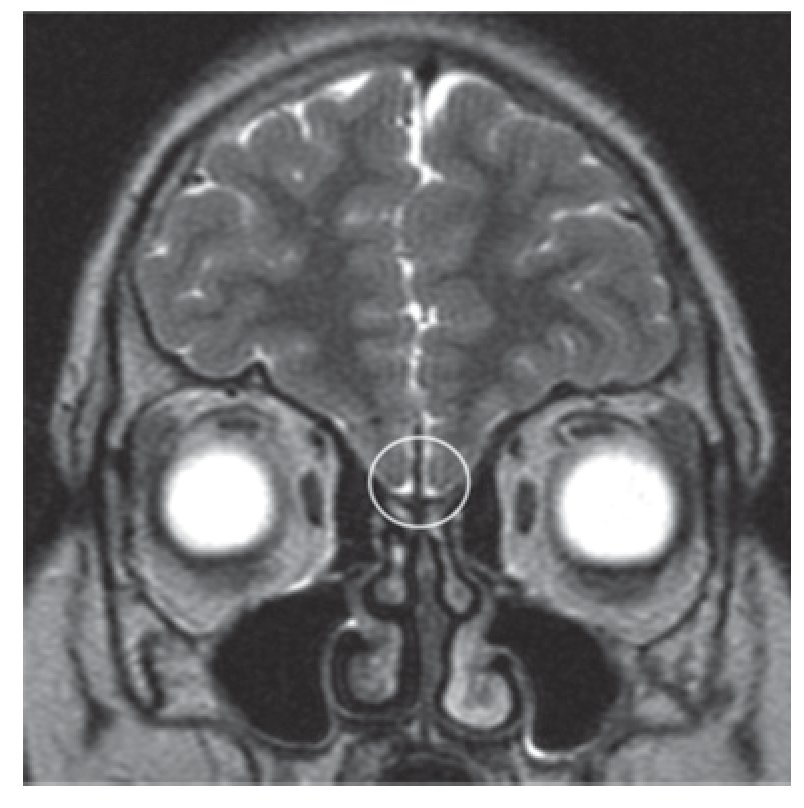
Figura 1. Ressonância magnética cerebral do caso 1: bolbos olfativos não visíveis (área assinalada).
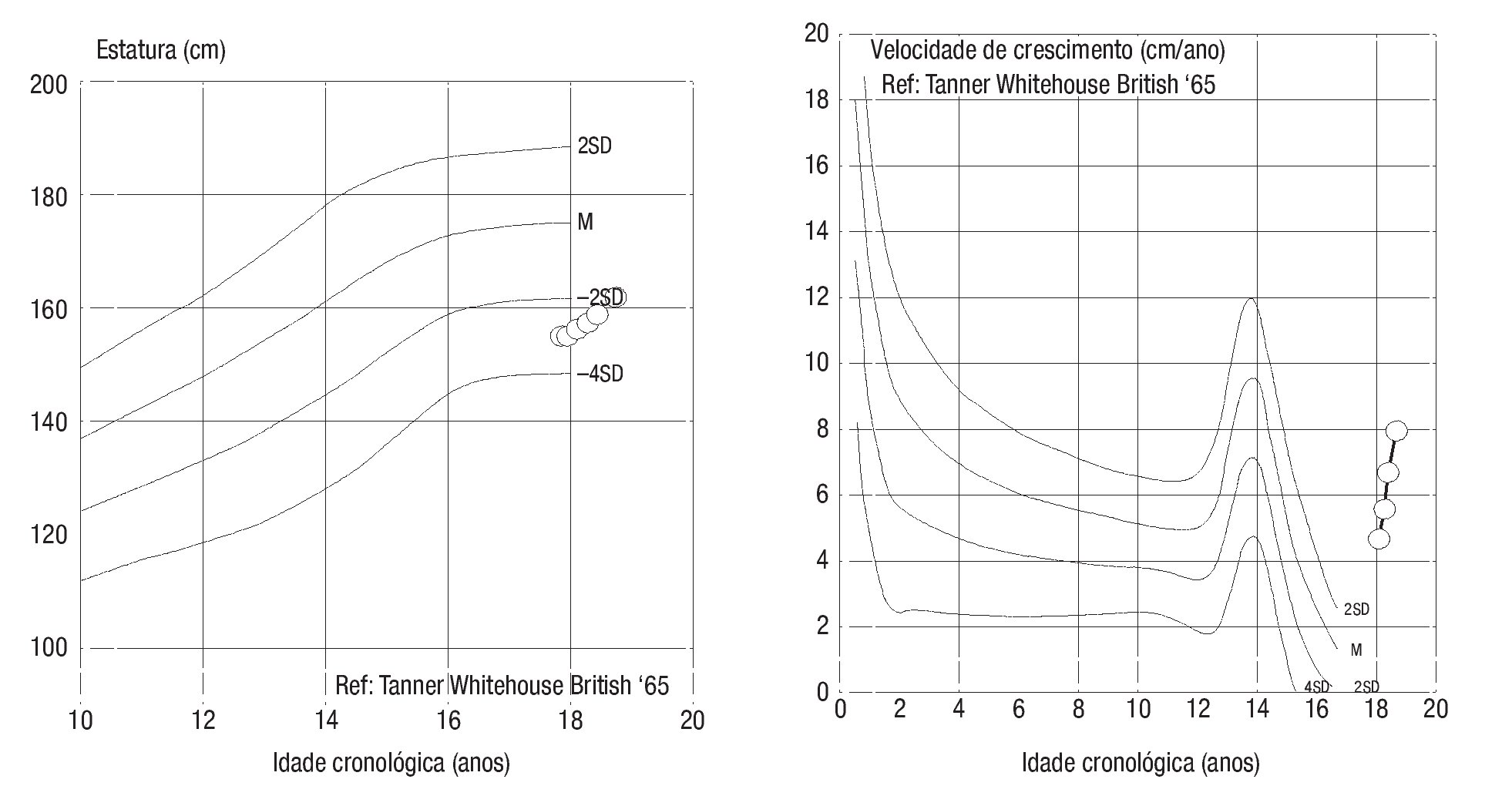
Figura 2. Evolução da curva estatural e da velocidade de crescimento do caso 1 desde a instituição da terapêutica.
Caso 2
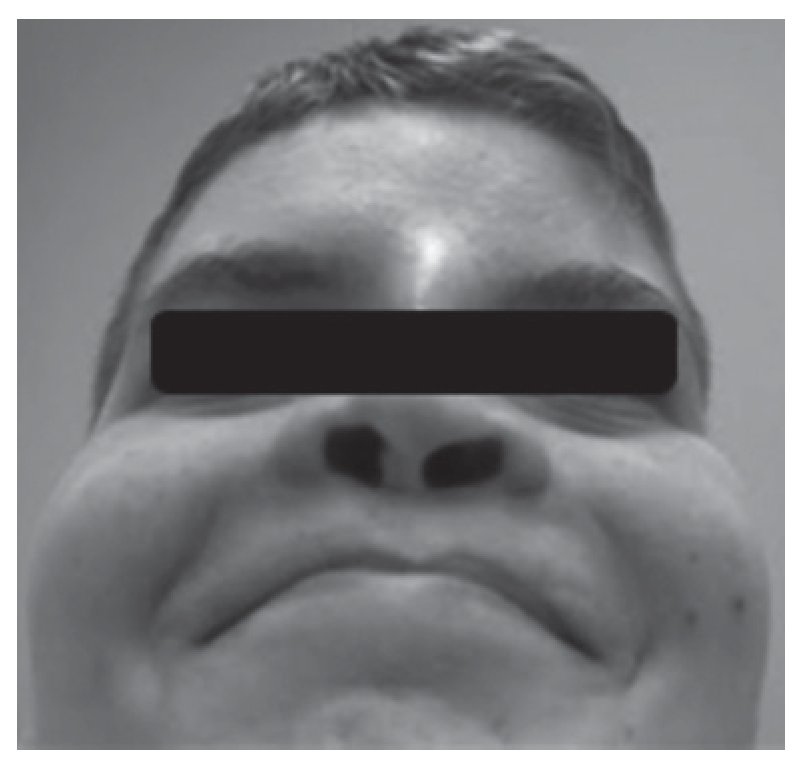
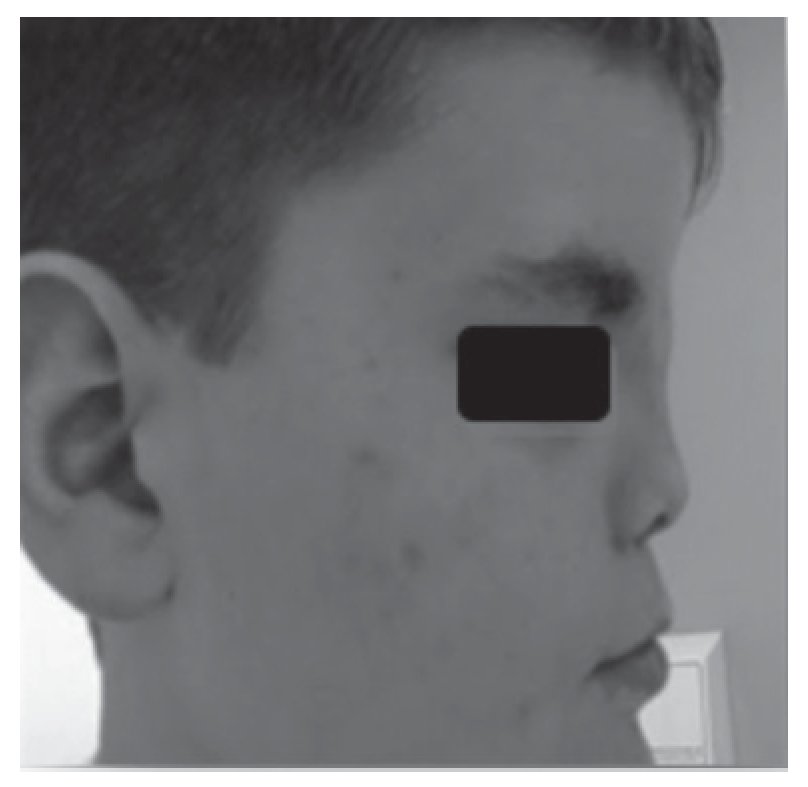
Adolescente do sexo masculino referenciado à consulta aos 16 anos e 6 meses por atraso pubertário. Foi submetido a cirurgia por lábio leporino (Figura 3) no primeiro ano de vida e era seguido em consulta de cirurgia plástica por dismorfia craniofacial (achatamento dos ossos da face na região fronto-nasal - Figura 4) e em consulta de neurologia por epilepsia associada a malformação do hemisfério cerebral esquerdo. Embora nunca a tivesse valorizado, referia diminuição da sensibilidade olfativa. Negava antecedentes familiares de anosmia, atraso pubertário ou infertilidade. Na primeira avaliação apresentava estatura de 182 cm (percentil 89), pénis críptico, pilosidade púbica Tanner 1, testículo esquerdo < 4 ml na bolsa esquerda e testículo direito não palpável. A estatura alvo era desconhecida - tratava-se de uma família monoparental com pai de identidade desconhecida. Dos exames auxiliares de diagnóstico efetuados salienta-se: prolactina e função tiroideia normais, FSH: 0,2 mUI/ml, LH < 0,1 mUI/ml e testosterona total: 10 ng/dl. A idade óssea era sobreponível à idade cronológica. A ecografia escrotal revelou presença de testículo esquerdo na bolsa escrotal e do testículo direito no canal inguino-escrotal, ambos com dimensões inferiores às esperadas para o grupo etário (17 x 8 x 9 mm e 14 x 6 x 8 mm, respetivamente). Na RM cerebral era descrita a presença de um hemisfério cerebral esquerdo pequeno associado a extensa anomalia do desenvolvimento cortical do tipo polimicrogírico e heterotopia subcortical prolongada até à parede do ventrículo lateral, condicionando apagamento do corno frontal do mesmo e deformação da cabeça do núcleo caudado. Não eram visíveis os sulcos nem os bolbos olfativos. A glândula pituitária e o hipotálamo não apresentavam alterações de tamanho ou configuração nem lesões expansivas. O estudo genético está atualmente em curso. A ecografia reno-pélvica não evidenciou alterações. Iniciou testosterona intramuscular aos 16 anos e 7 meses com aumento progressivo da dose e boa evolução clínica. Seis meses após início da terapêutica surgiu discreto pelo facial, o estádio pubertário progrediu apresentando pénis com 10 cm de comprimento, pilosidade púbica Tanner 4 e volume testicular de 6 ml à esquerda. O testículo direito mantém-se hipoplásico.
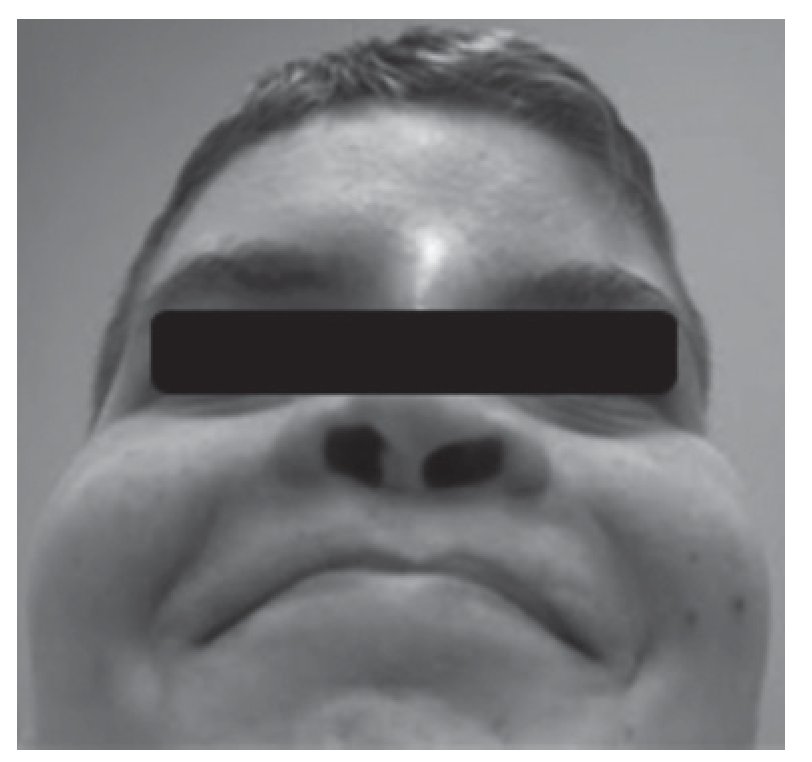
Figura 3. Cicatriz de cirrurgia após correção do lábio leporino (caso 2).
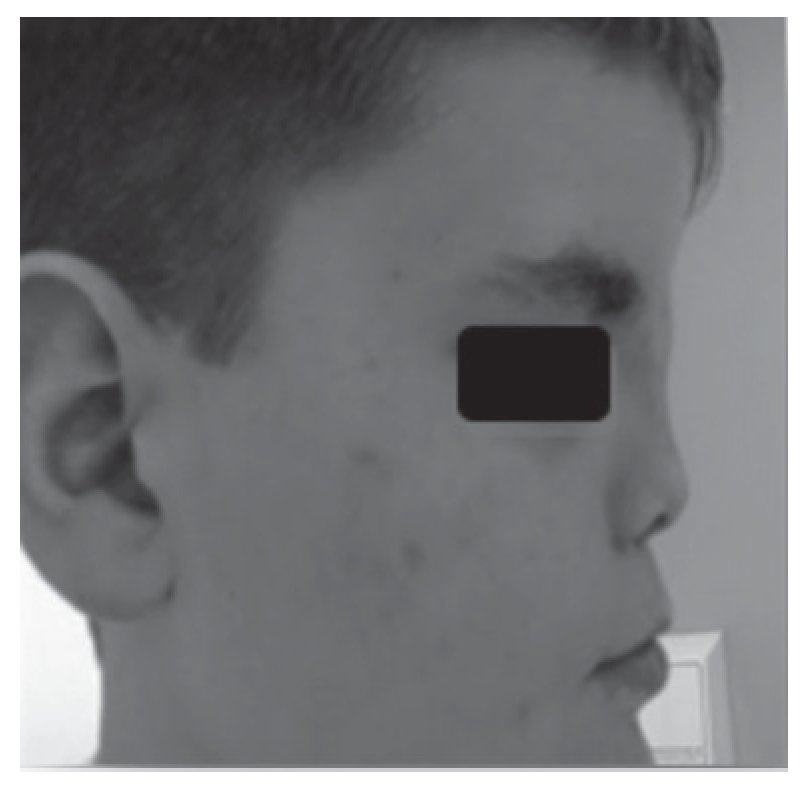
Figura 4. Hipoplasia óssea da região frontonasal (caso 2).
Caso 3
Adolescente do sexo feminino referenciada à consulta de endocrinologia pediátrica aos 12 anos por obesidade. Apresentava como antecedentes relevantes um atraso do desenvolvimento psicomotor e uma surdez neurossensorial bilateral com colocação bilateral de próteses auditivas aos 5 anos. As tias paternas tinham história de menarca tardia (aos 15 e 17 anos), mas não havia outros antecedentes familiares relevantes. Na primeira avaliação apresen--tava uma obesidade com índice de massa corporal de 26,9 kg/m2 (SD 3,43 percentil > 99,9), pilosidade púbica Tanner 3, adipomastia e acantose nigricans axilar e cervical. Apresentava critérios laboratoriais de insulinoresistência mas sem outras comorbilidades associadas à obesidade. Aos 16 anos de idade foi efetuada investigação por amenorreia primária. Apresentava nessa altura pilosidade púbica Tanner 4, desenvolvimento mamário Tanner 2, estatura de 155 cm (SD: -1,19; percentil 11) para uma estatura alvo de 153,5 cm. O cariótipo, previamente efetuado no contexto do atraso do desenvolvimento psicomotor, era normal (46, XX). A idade óssea aos 16 anos e 3 meses de idade cronológica era de 15 anos. Os níveis de gonadotrofinas e estradiol encontravam-se diminuídos (LH: 0,1 mUI/ml; FSH: 0,1 mUI/ml; estradiol: 17,8 pg/ml) e a função tiroideia e prolactina revelaram-se normais. Na ecografia pélvica era visível um útero de características e dimensões pré-púberes e a ecografia mamária mostrou predomínio adiposo com escassa matriz fibro-glandular. Na RM cerebral era descrita uma fóvea etmoidal hipoplásica, não se identificando os bolbos olfativos (possivelmente hipoplásicos) nem a porção anterior dos sulcos homónimos. Não foram observadas alterações da hipófise ou do hipotálamo. O estudo genético está atualmente em curso. Iniciou reposição hormonal com estrogénios transdérmicos aos 16 anos e 6 meses com aumento crescente da dose de acordo com a resposta pubertária, com evolução favorável. Verificou-se um crescimento progressivo e regular das dimensões uterinas, sobretudo à custa do aumento do corpo uterino que adquiriu forma em pêra e foi invertida a relação corpo/colo uterinos, passando a ser 2:1. Dois anos após, adicionou-se progestagénios e a adolescente teve a primeira hemorragia de privação aos 18 anos e 6 meses de idade. Atualmente apresenta hemorragias de privação regulares, pilosidade púbica Tanner 5 e desenvolvimento mamário Tanner 4.
Discussão
O atraso pubertário pode condicionar perturbações emocionais/ psíquicas e de adaptação social que podem ser irreversíveis3. Um período prolongado de baixos níveis de esteroides sexuais pode ainda comprometer a mineralização óssea, tornando o estudo da densidade óssea um exame a ponderar quando a reposição hormonal se inicia tardiamente5,13. O diagnóstico atempado torna-se assim imperativo. Contudo, o diagnóstico de SK é habitualmente efetuado na adolescência tardia ou idade adulta face à ausência do desenvolvimento pubertário5,8,11,13,14,17-19. O fenótipo de SK pode incluir diversas anomalias além das características típicas do hipogonadismo hipogonadotrófico e anosmia. O seu reconhecimento na lactância ou infância precoce pode levar a um diagnóstico e tratamento mais precoces5,8,11,13,14,16,19,20. Nos indivíduos do sexo masculino, a criptorquidia e o micropénis são achados frequentes no hipogonadismo hipogonadotrófico na lactância/infância precoce e estes achados estavam presentes nos casos 1 e 23,5,8,9,11,13,14,21.
A presença de micropénis foi reportada em 65% dos indivíduos com SK e a criptorquidia em 73% dos casos11, sendo frequentemente bilateral. Estes achados são mais frequentes em doentes com mutação no gene KAL1 e habitualmente associam-se a hipogonadismos graves21. Por outro lado, têm sido descritas diversas malformações associadas à SK. A anomalia renal mais frequente é a agenesia renal, detetada no caso 1. Outras malformações renais incluem rins em ferradura, mal-rotação renal, rim multiquístico e refluxo vesicoureteral11. O gene KAL1 foi o único associado a malformações renais, encontradas em 30% dos casos com mutação neste gene11,13.
Outras malformações incluem anomalias da linha média. Neste grupo, são particularmente frequentes o lábio leporino, presente no caso 2 descrito, e fenda de palato5,11,13,14 que ocorrem em 13 a 14% dos doentes e que estão habitualmente associadas a mutações dos genes FGR1, FGF8 e CHD711,13. Outros achados incluem agenesia dentária, palato em ogiva ou outras formas de fusão anómala da face com assimetria facial, evidente também no caso 25,11,13,15. A surdez neurossensorial, presente no caso 3, tem uma incidência de 28% nestes doentes e pode ser unilateral ou bilateral. Tem sido descrita em doentes com mutações nos genes KAL1, FGFR1, FGF8, PROKR2 e CHD711,13. A sincinésia ocorre frequentemente, estando descrita em 80 a 85% dos casos por alguns autores e associada particularmente a mutações no gene KAL1 embora raramente possa encontrar-se em mutações nos genes FGFR1, PROK2 e PROKR211,13. Mais raramente, podem associar-se anomalias músculo-esqueléticas (agenesia de um ou mais dedos13,21, hiperlaxidez11,13, metacarpos curtos, pé cavo5,13, braquidactilia5,13, sindactilia13, clinodactilia11,13, camptodactilia11,13 e pectus escavatum5,13), alterações da oculomotricidade (ptose, nistagmo11 e hipertelorismo5,13), alterações neurológicas (ataxia cerebelar9,11, agenesia corpo caloso5,13, paraplegia espástica19, atraso mental5,7,13,14 e epilepsia7,13,21), malformações cardíacas (comunicação interauricular ou interventricular, arco aórtico direito e transposição dos grandes vasos11) e obesidade mórbida5,13,14.
Nos 3 casos clínicos apresentados o diagnóstico foi efetuado por atraso pubertário na adolescência tardia. No hipogonadismo hipogonadotrófico congénito no sexo masculino, o início da reposição hormonal está recomendado quando a idade cronológica e/ou a idade óssea são superiores a 14 e 12 anos, respetivamente. No sexo feminino, recomenda-se iniciar o tratamento a partir dos 13 anos de idade cronológica ou 12 anos de idade óssea. Face a estas recomendações, nos 3 casos clínicos houve um atraso no início da reposição hormonal em resultado do diagnóstico tardio. Contudo, como pudemos constatar, em todos eles existiam indícios que poderiam ter levado a um diagnóstico mais precoce. Embora todos eles representem achados inespecíficos, quando associados a atraso pubertário, deve ser considerada e investigada a hipótese diagnóstica de SK.
É importante ainda relembrar que embora existam formas graves, em alguns indivíduos é possível existir um desenvolvimento parcial dos carateres sexuais secundários, com consequente atraso no diagnóstico5,8,13,14,20,21. Além disso, e tendo em conta que a maturação adrenal ocorre normalmente, os baixos níveis de
androgénios produzidos pela suprarrenal podem ser suficientes para o aparecimento da pubarca na idade prevista camuflando o atraso pubertário5,8,13,19. No caso 3, a pubarca e o aparecimento de botão mamário foram em parte responsáveis pela investigação ter sido apenas efetuada aos 16 anos em contexto da amenorreia primária. Além disso, a história de menarca tardia em duas tias paternas colocou como viável a hipótese de atraso constitucional do crescimento e maturação. A ocorrência de atraso constitucional do crescimento e maturação é, no entanto, mais frequente em familiares de doentes com SK do que na população em geral e este facto deve ser tomado em consideração5,7,14,15.
O caso 1 tem a particularidade de apresentar uma baixa estatura na primeira avaliação. Este achado não é habitual na SK. Apesar de estar associado habitualmente a um atraso da maturação esquelética, a taxa de crescimento linear é habitualmente normal, exceto pelo facto de não existir o surto de crescimento típico da puberdade2,3,5,13. Contudo, os esteróides sexuais têm um papel fundamental no crescimento nesta faixa etária e verificou-se, com a administração de testosterona intramuscular, uma normalização da antropometria com atingimento da estatura alvo. Assim sendo, parece-nos incontestável a hipótese de que o défice de testosterona era o elemento responsável pela baixa estatura.
A monitorização do volume testicular, do desenvolvimento mamário e dos restantes carateres sexuais secundários e a ecografia pélvica mostraram uma resposta favorável ao tratamento nos três casos clínicos apresentados.
Os autores consideram fundamental que numa situação de hipogonadismo hipogonadotróf ico, para além do exame físico cuidadoso, sejam explorados minuciosamente todos os antecedentes pessoais tendo em conta a panóplia de malformações que podem coexistir na SK e considerando ainda que a anosmia é frequentemente desvalorizada pelos doentes.
* Autor para correspondência.
Correio electrónico: marsandrina@gmail.com (S. Martins).
INFORMAÇÃO DO ARTIGO
História do artigo:
Recebido a 16 de janeiro de 2012
Aceite a 4 de julho de 2012
